高效液相色谱-串联质谱法同时测定面粉中7种非法添加剂
作者: 王颖怡 吴玉田 邹璐 孟春杨 钟雪 周贻兵
摘 要:目的:建立一种分析面粉中7种非法添加剂的高效液相色谱-串联质谱检测方法。方法:试样经乙腈∶水(1∶1,V∶V)提取、超声、离心、氮吹浓缩后,经Agilent Eclipse Plus C18 RRHD色谱柱分离、0.1%甲酸-5 mmol·L-1甲酸铵水和甲醇为流动相进行等度洗脱,采用多反应监测模式分析。结果:7种目标化合物在0.10~8.00 ng·mL-1呈良好的线性关系(R2>0.990),检出限为0.03~0.15 mg·kg-1,定量限为0.10~0.45 mg·kg-1;回收率在88.6%~104.3%,相对标准偏差在2.1%~5.8%(n=6)。结论:该方法简便、灵敏、准确,可满足小麦粉中7种非法添加物的同时检测要求。
关键词:高效液相色谱-串联质谱法;小麦粉;非法添加剂;噻苯咪唑;苯甲羟肟酸
Simultaneous Determination of 7 Illegal Additives in Flour by High Performance Liquid Chromatography-Tandem Mass Spectrometry
Abstract: Objective: To establish a high performance liquid chromatography-tandem mass spectrometry method for the determination of 7 illegal additives in flour. Method: The samples were extracted by acetonitrile∶water (1∶1,V∶V), concentrated by ultrasound, centrifugation and nitrogen blowing, separated by Agilent Eclipse Plus C18 RRHD column, and then eluted with 0.1% formic acid -5 mmol·L-1 ammonium formate water and methanol as mobile phase, and analyzed by multi-reaction monitoring mode. Result: There was a good linear relationship (R2>0.990) between 0.10~8.00 ng·mL-1 for the 7 target compounds. The limit of detection was 0.03~0.15 mg·kg-1, and the limit of quantification was 0.10~0.45 mg·kg-1. The recoveries were 88.6% ~104.3%, and the relative standard deviations were 2.1%~5.8%(n=6). Conclusion: The method is simple, sensitive and accurate, and can satisfy the simultaneous detection of 7 illegal additives in wheat flour.
Keywords: high performance liquid chromatography tandem mass spectrometry; wheat flour; illegal additives; thiobenzimidazole; benzohydroxamic acid
国内三大粮食作物之一的小麦是仅次于稻谷的第二大粮食作物,其含有大量碳水化合物和蛋白质,小麦粉(面粉)是其主要加工产品。国家统计局发布的资料表明,伴随着国内小麦出产量的不断增加,面粉的产量与消费量也在不断增加,但其安全性与品质也曾出现过严重的问题[1]。随着经济增长与生活水平的提升,市场对面粉类产品的品质有着越来越严苛的标准[2]。部分生产者为了追求最大利益,利用化学制剂提高面粉质量,掩盖其存在的质量问题,如使用硫脲作为增白剂、曲酸作为保鲜剂,以及使用噻二唑作为抗生素等非食品原料。此外,如果贮存环境或条件达不到国家规定的要求,小麦在贮藏期间易发霉变质,或者受到昆虫与老鼠啃食,从而造成面粉污染。目前,不法商家为了避免小麦粉出现醒发、复色和黑点,向其中非法添入了苯甲羟肟酸以及三聚硫氰酸三钠盐成分,最终使产品达到出厂标准。上述行为不仅给消费者的身体健康带来很大危害,还会对食品安全产生影响。原国家食药监管总局2017年公布的《总局关于进一步加强小麦粉质量安全监管的公告》的第4条内容针对企业的生产过程明确规定,严格禁止将非食品原材料添入面粉中,如过氧化苯甲酰、次磷酸钠、硫脲、间苯二酚、过硫酸盐、噻二唑和曲酸等[3]。因此,在使用添加剂时,需坚持“安全、合理、有限、有效”的原则,严格执行国家相关规定。
当前,国内与国际上针对上述非法添加剂主要采用高效液相色谱法[4]、高效液相色谱-串联质谱法[5-6]、近红外光谱法[7]和电化学方法[8-9]等多种检测手段。申科敏等[4]通过HPLC法测定小麦粉中的曲酸、噻二唑、硫脲等化学成分;国振等[10]建立了高效液相色谱-二极管阵列检测器同时测定小麦粉及面粉处理剂中的4种违法添加剂的方法,以上两种方法的检出限均偏高;林芳[6]以纯水溶液为提取剂,超声提取,建立超高效液相色谱串联质谱法同时测定面粉中曲酸和噻二唑,虽然HPLC-MS/MS填补了曲酸和噻二唑检测方法的空白,但在实际样本筛查中需要进一步完善。除此之外,同时测定食品中噻苯咪唑和四环素等成分含量的报道仍然较少。因此,本研究采用HPLC-MS/MS筛查确认样品中的硫脲、曲酸、噻苯咪唑、噻二唑、四环素、苯甲羟肟酸和三聚硫氰酸三钠盐7种化合物,通过含量检测分析,了解市售小麦粉非法添加剂的污染水平,为食品安全监管提供有效手段和技术支持,全面保障小麦粉的安全。
1 材料与方法
1.1 材料与试剂
样品为2022年贵阳市市售小麦粉样品。
噻二唑(纯度>99.6%),北京振翔科技有限公司;四环素(纯度>95.5%)、三聚硫氰酸三钠盐(纯度>98.3%)、硫脲(纯度>99.2%)、苯甲基羟肟酸(纯度>99.9%)、曲酸(纯度>98.3%)和噻苯咪唑(纯度>99.9%),上海安谱实验科技股份有限公司;乙腈和甲醇,美国飞世尔试剂公司。
1.2 仪器与设备
Agilent 6470三重四极杆串联质谱仪,美国Agilent公司;Agilent 1290超高效液相色谱仪,美国Agilent公司;Milli-Q超纯水设备,美国Merck;3-30KS台式高速冷冻离心机,德国Sigma;MS3漩涡混匀设备,德国IKA;T460 H超声波清洗设备,北京博劢行仪器。
1.3 前处理方法
称取均质试样5 g,于已备好的50 mL聚丙烯离心管中,加入5 mL水涡旋混匀后加入5 mL乙腈,漩涡混合30 s,超声提取20 min,10 000 r·min-1离心10 min后(处理温度小于5 ℃),移取1 mL上清液于离心管中,氮吹至近干即可。加入流动相
1 mL,经过涡旋混合均匀后,经0.22 µm有机微孔滤膜过滤,待上机。
1.4 仪器条件
1.4.1 色谱条件
Agilent Eclipse Plus C18 RRHD色谱柱(3.0 mm×150 mm,1.8 μm);流动相:A为0.1%甲酸-5 mmol·L-1甲酸铵水溶液,B为甲醇溶液,A+B=74+26;流速:0.3 mL·min-1;柱温:40 ℃;进样量:5 μL;等度洗脱。
1.4.2 质谱条件
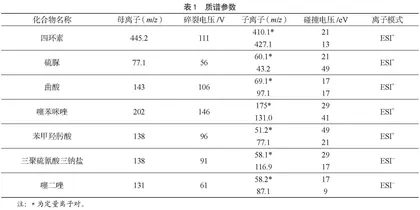
离子源模式:电喷雾离子源(ESI);正模式喷雾电压:3 000 V;负模式喷雾电压:2 500 V;鞘气温度:250 ℃;脱溶剂温度:300 ℃;喷雾器:35 psi;鞘气流速:11 L·min-1;采集模式:多反应监测MRM模式。化合物质谱参数见表1。
2 结果与分析
2.1 质谱及色谱条件的优化
化合物中的吸电子基团具有高度亲电性,能够吸引周围电子,从而使H+易于生成[M+H]+类型的离子峰。例如,硫脲、噻苯咪唑结构中硫原子和氮原子上的孤对电子使得硫脲分子具有亲电性,而苯甲羟肟酸、曲酸及四环素结构中含有酰胺、羰基等极性官能团,具有中等亲电性。所以,上述化合物都采用了电喷雾离子正离子模式。由于噻二唑及三聚硫氰酸三钠盐具有杂环结构,电子云密度分布不均匀,使其容易丢失H+,从而产生[M-H]-型离子峰。因此,试验拟采用电喷雾离子负离子模式。分别配制浓度均为500 ng·mL-1的7种化合物的标准溶液,在ESI+和ESI-模式下对目标物进行全扫描,通过对准分子离子的信号响应进行筛选,改善碎裂电压和碰撞能量,获得最佳的母离子丰度。在此基础上,通过二级质谱扫描,筛选出两个具有代表性的碎片离子,并对碰撞电压进行改善,将丰度较高的子离子选择为定量离子。
在优化色谱条件中,探究不同流动相(甲醇-水溶液、乙腈-水溶液、0.1%甲酸水-乙腈溶液、0.1%甲酸-5 mmol·L-1甲酸铵水-甲醇溶液)对7种目标检测物的分离情况。结果表明,使用甲醇-水溶液、乙腈-水溶液时,四环素峰发生峰拖尾现象;当流动相为0.1%甲酸水-乙腈溶液时,硫脲、三聚硫氰酸三钠盐的色谱峰形态发生分叉。因此,为改善峰形及分离度,试验最终选择0.1%甲酸-5 mmol·L-1甲酸铵水-甲醇溶液作为流动相,采用Agilent Eclipse Plus C18 RRHD(3.0 mm×150 mm)色谱柱,在等度洗脱方式下7种检测物分离效果良好。
2.2 提取条件的优化
鉴于硫脲、曲酸和苯甲羟肟酸的强极性,分别研究了水、甲醇、甲醇-水(1∶1,V∶V)、乙腈以及乙腈-水(1∶1,V∶V)对化合物提取率的影响。结果表明,以水为溶剂进行提取试验时,其提取强极性成分的效果最好,而对极性比较弱的噻二唑和噻苯咪唑则反之;而甲醇作为提取液时,曲酸及硫脲会产生严重的溶剂效应,如果利用甲醇-水(1∶1,V∶V)为溶剂进行提取,样品溶液浑浊且呈凝胶状,无法进行后续实验;采用纯乙腈溶液进行提取时,化合物的极性越大,提取率越低。相反极性相对较低的化合物如苯甲羟肟酸、噻二唑、噻苯咪唑等,其提取率均有所增加。综上,本研究选择水-乙腈(1∶1,V∶V)作为提取溶剂,确保色谱峰峰形的正常。
2.3 基质效应
在液质分析中,食品样品中的内源性成分及样品前处理过程中的外源性成分会影响目标物的离子化效率,从而导致基质效应的产生,常表现为基质增强或基质抑制。在此基础上,使用提取后的空白样品溶液进行加标,可评价化合物的基质效应(Matrix Effect,ME),依据基质效应数学模型进行评估,即ME=Y/X×100%。其中,Y为空白样品基质中标准物质的响应均值,X为纯溶剂中添加等量标准物质的响应均值[11-12]。结果表明,噻二唑、四环素、三聚硫氰酸三钠盐、硫脲、苯甲羟肟酸、曲酸和噻苯咪唑这7种目标分析物表现出较弱的基质效应并在可接受范围,其结果分别为87%、110%、83%、91%、96%、105%和102%。为减少基质效应对定量分析的干扰,本文的定量方法使用基质匹配标准曲线法。
2.4 线性范围、检出限、定量限
根据1.3样品前处理步骤,采用基质匹配标准曲线法,将7种违禁添加剂配制为一系列不同浓度的混合标准液,于最佳色谱和质谱条件下进样,并绘制标准曲线。与此同时,本实验通过在空白基质中添加低质量浓度混合标准溶液,按以上方法进行测定,经响应值3倍和10倍的信噪比计算检出限和定量限。由表2可知,7个目标物均存在很好的线性关系(R2>0.99)。