液相色谱-串联质谱法同时检测鱼中氯霉素类和硝基咪唑类药物残留
作者: 勇艳华
摘 要:本实验建立了同时检测鱼肉中3种氯霉素类和5种硝基咪唑类药物残留的液相色谱-串联质谱方法。采用乙酸乙酯提取,提取液浓缩后纯水定容,正己烷脱脂,在优化的液相色谱条件下,3种氯霉素类和5种硝基咪唑类药物在8 min内实现良好分离。氯霉素类检出限均为0.1 µg/kg,甲硝唑检出限为0.5 µg/kg,其他硝基咪唑类均为1.0 µg/kg。在线性范围(氯霉素类0.5~50.0 μg/L,硝基咪唑类2.0~100.0 μg/L)内,相关性良好,相关系数r>0.996 0,空白基质3个添加水平的平均回收率为82.3%~109.0%,相对标准偏差(n=6)为3.6%~12.0%。本方法经济、快速、简单和高效,能同时满足鱼肉中3种氯霉素类和5种硝基咪唑类药物残留的检测要求。
关键词:鱼肉;氯霉素类;硝基咪唑类;药物残留;液相色谱-串联质谱仪
LC-MS/MS Simultaneous Determination of Chloramphenicol and Nitroimidazole Residues in Fish
YONG Yanhua
(Dalian Center for Certification and Food and Drug Control, Dalian 116630, China)
Abstract: A high performance liquid chromatography-tandem mass spectrometry method for simultaneous determination of 3 chloramphenicol and 5 nitroimidazole residues in fish was established. The extract was extracted with ethyl acetate, concentrated with pure water and degreased with n-hexane, under the optimized conditions of liquid chromatography, the 3 chloramphenicol and 5 nitroimidazole residues were separated well within 8 min. The detection limits of chloramphenicol, metronidazole and other nitroimidazole were 0.1 µg/kg, 0.5 µg/kg and 1.0 µg/kg respectively. In the linear range (chloramphenicol 0.5~50.0 μg/L, nitroimidazole 2.0~100.0 μg/L), the correlation was good, and the linear correlation coefficient was r>0.996 0. The average recoveries were 82.3%~109.0% with relative standard deviations (n=6) of 3.6%~12.0% at 3 supplemental levels. The method is economical, rapid, simple and efficient, and this method can fully meet the requirement of simultaneous determination of 3 chloramphenicol and 5 nitroimidazole residues in fish.
Keywords: fish; chloramphenicol; nitroimidazole; drug residue; liquid chromatography-tandem mass
氯霉素类药物为广谱抗生素,具有良好的抗菌能力,被广泛应用于水产养殖。氯霉素类药物的毒副作用主要表现为对血液系统造成的毒性。硝基咪唑类药物有致突变性和潜在的致癌性,很多国家已将其列为违禁药物。欧盟在20世纪90年代已经禁止洛硝哒唑、地美硝唑和甲硝唑用于可食用动物,2002年美国食品与药物管理局又公布了11种禁止在进口动物源性食品中使用的药物名单,其中包括硝基咪唑类药物。我国农业部和国家药品监督管理局第227号公告中规定,甲硝唑、地美硝唑及其盐、酯及制剂不允许以促进动物生长为目的在所有食用动物饲养过程中使用。农业部公告第235号文件规定,水产品中氯霉素、甲硝唑、地美硝唑、洛硝达唑、羟基甲硝唑和羟甲基甲硝唑均不得检出,但是甲砜霉素在鱼肉中限量50 μg/kg,氟苯尼考在鱼肉中限量1 000 μg/kg、在其他动物肌肉中为100 μg/kg。
氯霉素类药物的检测方法有很多,分别为酶联免疫法、液相色谱法、气相色谱法、气相色谱串联质谱法和液相色谱-串联质谱法,现在普遍使用液相色谱串联质谱法[1-11]。检测硝基咪唑类及其代谢物的方法有酶联免疫法、高效液相色谱法,主要采用液相色谱-串联质谱法[12-18]。但同时检测水产品中氯霉素类和硝基咪唑类及其代谢物的报道较少,标准《出口动物源食品中多类禁用药物残留量检测方法 液相色谱-质谱/质谱法》(SN/T 3235—2012)中虽然包含了氯霉素和5种硝基咪唑类物质,但未包含甲砜霉素和氟甲砜霉素,且需碱化乙腈、酸化乙腈各提取1遍,略显烦琐,此外使用的提取试剂乙腈毒性较强[19]。本文建立了同时检测氯霉素族和硝基咪唑类药物残留的液相色谱-串联质谱分析方法,采用毒性较小的乙酸乙酯为提取试剂,进行液液萃取,正己烷脱脂,检出限可达到要求,重复性好、准确度高、操作简便,可满足鱼肉中两类药物残留的检测要求,适合用于生产、流通、餐饮及进出口环节的质量监管。
1 材料与方法
1.1 仪器与试剂
液相色谱-串联质谱仪(1290/6495,美国Agilent公司);均质机(德国IKA公司);旋转蒸发仪(瑞士Buchi公司);CF16RXII立式大容量高速离心机(日本Hitachi公司);赛多利斯全自动超纯水机(德国Sartorius公司);Agilent poroshell 120 EC-C18色谱柱(4.6 mm×50.0 mm,2.7 μm,美国Agilent公司)。
实验中甲醇、乙酸乙酯和正己烷均为色谱纯;实验用水为超纯水;无水硫酸钠为分析纯;甲硝唑、地美硝唑、洛硝达唑、羟基甲硝唑、羟甲基甲硝咪唑、羟甲基甲硝咪唑-D3、氯霉素、氯霉素-D5、氟甲砜霉素和甲砜霉素均为Dr.E公司生产,纯度大于98%。
1.2 标准溶液的配制
氯霉素类:分别称取氯霉素、氟甲砜霉素和甲砜霉素标准品约0.010 0 g(精确至0.000 1 g)于10 mL容量瓶中,甲醇溶解并定容至刻度,得浓度为1.0 mg/mL的标准储备液。准确吸取各标准储备液0.1 mL于100 mL容量瓶中,甲醇定容至刻度,得浓度为1 μg/mL的混合标准溶液。硝基咪唑类:分别称取甲硝唑、地美硝唑、洛硝达唑、羟基甲硝唑和羟甲基甲硝咪唑标准品约0.010 0 g(精确至0.000 1 g)于10 mL容量瓶中,甲醇溶解并定容至刻度,得浓度为1.0 mg/mL的标准储备液。准确吸取各标准储备液0.1 mL于100 mL容量瓶中,甲醇定容至刻度,得浓度为1 μg/mL的混合标准溶液。氘代内标:分别称取氯霉素-D5和羟甲基甲硝咪唑-D3标准品0.010 0 g(精确至0.000 1 g)于100 mL容量瓶中,甲醇溶解并定容至刻度,得浓度为100 μg/mL的内标标准储备液。准确吸取内标标准储备液各0.1 mL分别置于100 mL容量瓶中,甲醇定容至刻度,得浓度为100 μg/L的内标标准溶液。
取适量氯霉素类、硝基咪唑类混合标准溶液和内标标准溶液,用20%甲醇逐级稀释至氯霉素类的浓度为0.5 μg/L、2.0 μg/L、5.0 μg/L、20.0 μg/L、50.0 μg/L;硝基咪唑类的浓度为2.0 μg/L、10.0 μg/L、20.0 μg/L、60.0 μg/L和100.0 μg/L;内含5.0 μg/L氯霉素-D5和20.0 μg/L羟甲基甲硝咪唑-D3。
1.3 样品前处理
称取5.00 g(精确到0.01 g)混合均匀的样品于50 mL离心管中,加入20 μL羟甲基甲硝咪唑-D3标准中间液、5 μL氯霉素-D5标准中间液、25 mL乙酸乙酯及适量无水硫酸钠,均质1 min,12 000 r/min高速离心,将上清液转移至圆底烧瓶中,再加入25 mL乙酸乙酯重复提取1次,合并提取液至圆底烧瓶中,减压旋转蒸发至近干。1 mL纯水定容,2 mL正己烷脱脂,涡旋1 min,取水层过0.2 µm微孔有机滤膜,待测定。
1.4 仪器条件
1.4.1 色谱条件
色谱柱:Agilent poroshell 120 EC-C18;柱温:30 ℃;流动相:A为纯甲醇,B为高纯水;梯度洗脱程序:0~1.5 min(25%A),1.5~4.1 min(25%~100%A);4.1~6.5 min(100%A);6.5~6.6 min(25%A);流速:0.3 mL/min;进样体积:5 μL。
1.4.2 质谱参数
电喷雾离子源(Electron Spray Ionization,ESI);
扫描方式:正负同时扫描;毛细管电压:3 500 V;
监测方式:多反应监测(Multiple Reaction Monitoring,
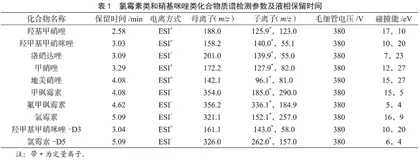
MRM);离子源温度:200 ℃;去溶剂气温度:300 ℃;去溶剂气:氮气13 L/min;锥孔气:氮气12 L/min。其他参数详见表1。
2 结果与分析
2.1 提取试剂的选择
利用空白基质加标考察回收率,对提取试剂乙腈、甲醇和乙酸乙酯进行筛选,实验结果表明,3者在回收率上无明显差异,乙酸乙酯毒性较小,确定其为本实验的提取试剂。提取后,用正己烷脱脂,可提高回收率。
2.2 流动相的选择
氯霉素类药物和硝基咪唑类的结构决定了其可以在C18色谱上获得分离。本实验探讨了甲醇和乙腈对目标化合物的洗脱效果,实验结果表明,两者均能较好地分离表1中的10种物质,但考虑到价格和毒性等因素,最终选择甲醇;氯霉素类为负模式采集,酸抑制负模式的电离,影响灵敏度,因此最终确定流动相为纯甲醇和高纯水。在1.4.1所述梯度洗脱条件下,在8 min内可获得这10种物质的最佳峰形,达到快速分析的目的。
2.3 线性范围及检出限
氯霉素类和硝基咪唑类分别在0.5~50.0 μg/L和2.0~100.0 μg/L浓度与峰面积成线性相关,线性方程及相关系数见表2。利用低浓度添加回收确定本方法氯霉素类检出限为0.1 µg/kg,硝基咪唑类检出限中甲硝唑为0.5 µg/kg,其他均为1.0 µg/kg。
2.4 回收率和精密度
用空白鱼肉基质进行加标试验,采用标准曲线内标法计算测定结果,以测定值对理论添加量的百分比表示回收率。每个添加水平进行6次平行实验,测定值的相对标准偏差表示方法精密度,结果见表3。由表可知,8种物质的加标回收均能满足检验需要。
2.5 实际样品分析
利用本方法对市场销售的50余份鱼进行检测,其中,1份鱼肉中检出氯霉素0.32 µg/kg,1份鱼肉中检出甲硝唑5.21 µg/kg,其他样品均未检出。