超高效液相色谱-串联质谱同位素内标法测定羊肉中33种兽药残留
作者: 陈旭晋 曹佳 陆宇阳 苑华宁 章雪明 张小刚
摘要 [目的]建立超高效液相色谱-串联质谱同时测定羊肉中33种兽药残留的方法。[方法]样品用90%乙腈水(含2.0%甲酸)提取,通过Oasis Prime HLB净化,浓缩后用流动相定容。采用Waters ACQUITY UPLC HSS T3 柱(2.1 mm×100 mm,1.8 μm)分离。以甲醇和0.1%甲酸水为流动相梯度洗脱,在电喷雾离子源下正、负离子切换扫描,多反应监测(MRM)模式下,结合保留时间和特征离子信息,利用同位素内标法定量。[结果]33种兽药在0.5~40.0 ng/mL线性关系良好(r>0.99),最低检出浓度在0.08~0.10 ng/mL。加标回收率为83.7%~115.0%,RSD为0.58%~11.49%。[结论]该方法具有处理简单、灵敏度高、重复性好等优点,满足羊肉中33种兽药的定量检测。
关键词 超高效液相色谱-串联质谱;同位素内标;羊肉;兽药残留
中图分类号 S851.34+7 文献标识码 A 文章编号 0517-6611(2024)03-0195-07
doi:10.3969/j.issn.0517-6611.2024.03.047
Determination of 33 Veterinary Drug Residues in Mutton by Ultra High Performance Liquid Chromatography-Tandem Mass Spectrometry with Isotope Internal Standard Method
Abstract [Objective]To establish a method for the simultaneous determination of 33 kinds of veterinary drug residues in mutton by ultra high performance liquid chromatography-tandem mass spectrometry. [Method]The sample was extracted with 90% acetonitrile water (containing 2.0% formic acid), purified by Oasis Prime HLB, concentrated, and diluted with mobile phase. Then separated by Waters ACQUITY UPLC HSS T3 column(2.1 mm×100 mm,1.8 μm),using 0.1% formic acid and acetonitrile as mobile phase.Under electrospray ionization source, switching between positive and negative ion modes, in Multiple Reaction Monitoring (MRM) mode, utilizing the isotope internal standard method for quantification by combining retention time and characteristic ion information. [Result] The linear relationship of the 33 kinds of veterinary drugs was good in the concentration range of 0.5-40.0 ng/mL(r>0.99),and the lowest detection concentration was 0.08-0.10 ng/mL.Adding standard recovery rates was 83.7%-115.0%,the RSD was 0.58%-11.49%. [Conclusion]The method has the simple processing,high sensitivty and good reproducibility,and is suitable for the quantitative detection of 33 kinds of veterinary drug residues in mutton.
Key words UPLC-MS/MS;Isotope internal standard;Mutton;Veterinary drug residues
近年来,我国动物生产和兽药使用量的增加已引起人们对兽药残留问题的广泛关注。为控制动物产品中的兽药残留水平,我国已实施了多项措施,包括制定相关法规和标准,规定了最大残留限量,并实施了监测计划[1]。同时,对不遵守相关法规的个人或单位实施处罚。尽管如此,动物产品中仍然存在违禁兽药残留的情况[2-5],需要持续加强监管和控制措施。
兽药残留检测是发现动物组织中违禁兽药残留的有效手段。兽药残留检测面临着许多技术难题,包括受兽药种类、前处理方法、畜产品基质等因素的影响。其中,前处理方法复杂耗时、检测效率低以及缺乏多种类药物残留联检技术等问题尤为突出。随着液质联用仪的发展,动物源性产品中多种兽药残留的联合检测技术已成为研究热点。如徐伟等[6]利用WondaPak QuEChERS多兽药残留专用提取包和净化包对猪肉中125种兽药进行检测,20 min内完成前处理,灵敏度和重现性均满足日常检测要求;许均图等[7]利用Captiva EMR-Lipid净化检测鸡肉中12种兽药残留,检出限低且可以同时检测鸡肉中多类兽药残留;黄泽玮等[8]采用QuEChERS EMR-Lipid技术结合LC-MS/MS对猪肉中55种兽药残留进行快速的筛查和确证,55种化合物在30 min内完成分离,方法快速有效;李永琴等[9]通过Oasis PRIME HLB净化结合LC-MS/MS建立了对牛肉中12类80种兽药及其代谢物的快速检测方法,方法定量限在2.0~5.0 μg/kg,具有灵敏度高和重复性好的特点;张建伟等[10]建立了同时测定羊奶和牛奶中8大类43种药物的残留方法,样品经氨化乙腈和酸化乙腈水重复提取,低温冷冻净化除脂后,LC-MS/MS检测,方法灵敏度高、准确性好且适用性强。羊肉含有丰富的蛋白质、铁、锌、维生素B12等营养素,而且含有所有必需氨基酸。由于其独特的口感和营养价值,羊肉深受消费者的喜爱。目前多残留检测技术在羊肉基质上相关报道较少。
该研究旨在开发一种检测羊肉中多种兽药残留的方法,选择33种典型兽药,采用通过式的净化小柱结合LC-MS/MS技术进行检测,并通过同位素内标定量确保方法的准确性和可靠性。
1 材料与方法
1.1 试验材料
1.1.1 主要试剂和耗材。乙腈、甲醇、甲酸、乙酸铵(色谱纯,美国霍尼韦尔公司);试验用水为 Milli-Q超纯水(美国 Millipore);Oasis Prime HLB 固相萃取柱(200 mg/6 mL,美国Waters公司);Captiva EMR-Lipid固相萃取柱(300 mg/3 mL,美国Agilent公司);Anavo HMR-Lipid固相萃取柱(300 mg/3 mL,北京纳鸥科技有限公司)。
1.1.2 标准物质。喹诺酮类混标、酰胺醇类药物混标、金刚烷胺、环丙沙星-D8、恩诺沙星-D5、氯霉素-D5、盐酸金刚烷胺-D5(100 μg/mL,1 mL,天津阿尔塔科技有限公司);磺胺类药物混标、β-受体激动剂药物混标、磺胺类同位素混标、β-受体激动剂同位素混标(100 μg/mL,1 mL,上海安谱实验科技股份有限公司)。
1.1.3 主要仪器。QTRAP 5500串联四极杆质谱系统(美国SCIEX);H-Class超高效液相色谱串联质谱仪(美国Waters);GD 16高速研磨均质仪(深圳新锐精仪);XcelVap自动浓缩工作站(美国Horizon);5804R高速冷冻离心机(德国艾本德)。
1.2 试验方法
1.2.1 前处理方法。
称取样品5.00 g于50 mL具塞离心管中,加入适量同位素内标物,加入90%乙腈水(含2.0%甲酸)9 mL,均质提取3 min, 于离心机中10 000 r/min离心3 min。取3 mL上清液过Oasis Prime HLB 固相萃取柱,收集全部滤出液,涡旋混匀。45 ℃水浴氮吹浓缩至近干,用5%甲醇水溶液(含0.1%甲酸)定容至1 mL,过0.22 μm滤膜后上机。
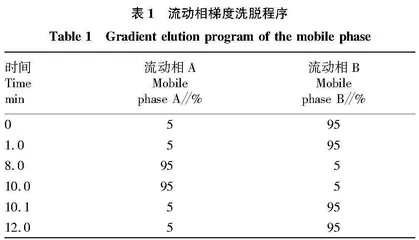
1.2.2 液相条件。色谱柱为ACQUITY UPLC HSS T3 柱(2.1 mm×100 mm,1.8 μm);柱温40 ℃;流速0.3 mL/min;流动相A为甲醇,流动相B为0.1%甲酸水;梯度洗脱程序见表1。
1.2.3 质谱条件。
离子源为电喷雾离子源(ESI)。多反应监测(MRM)。氯霉素、氟苯尼考、甲砜霉素为负离子扫描,其余化合物为正离子扫描。离子化电压正离子模式为5 500 V,负离子模式为4 500 V。离子源温度500 ℃。辅助加热气344.74 kPa。气帘气344.74 kPa。各化合物详细参数见表2。
1.2.4 基质混合标准工作液的制备。
取 7 份空白羊肉经前处理氮吹干后的残余物,依次加入 0.5、1.0、2.0、5.0、10.0、20.0、40.0 ng/mL的 33种药物混合标准溶液 1 mL,其中内标浓度为5.0 ng/mL完全溶解后,过0.22 μm滤膜,得到基质混合标准工作液。
2 结果与分析
2.1 流动相的选择
液相色谱-串联质谱的流动相体系一般为有机相加水相。有机相一般选择甲醇和乙腈,乙腈相较于甲醇来说洗脱能力更强,但对目标物和杂质之间分离效果不如甲醇[11]。水相一般为纯水或者添加适量的甲酸、乙酸铵。经测试,当有机相为乙腈时,33种兽药中,西马特罗有杂峰干扰。当有机相为甲醇时,西马特罗的目标峰和杂峰可以完全分离,因此选择甲醇为流动相。水相可以选择纯水或加入适量甲酸、乙酸铵,经测试,甲酸的加入可以改善喹诺酮类药物的峰型,而乙酸铵则会抑制部分兽药的响应。最终选择甲醇-水(含0.1%甲酸)作为流动相,所有33种兽药都能得到较好的分离。33种兽药选择离子流图如图1所示。
2.2 提取溶剂的选择
这些兽药化合物易溶于乙腈、甲醇等极性溶剂。当提取含有较高动物性蛋白、脂肪的样品时,使用极性强的甲醇提取液可能会导致一些问题,例如提取液浑浊、净化处理时易堵塞固相萃取柱,以及浓缩后瓶壁上会附着难以溶解的凝结物,如白色的蛋白质和脂肪。相比之下,乙腈对脂肪的溶解度较小,同时对蛋白质具有沉淀作用。在这种情况下,乙腈是更好的选择[12]。乙腈中加入甲酸或乙酸可以使提取环境呈酸性,有利于某些兽药在提取过程中的离子化程度,提高它们的溶解度和提取效率。虽然乙腈会使蛋白变性凝聚的效果较好,但同时可能会阻碍目标兽药的提取。因此,在乙腈中加入部分的水辅助提取能取得较好的效果[13]。该试验测试了不同浓度和不同含甲酸量的提取溶液,并统计了各类兽药在质谱上的平均峰面积。经测试发现(图2),提取液中水相比例的增加会影响某些兽药的提取效率,而甲酸的含量同样会影响各类兽药的响应,最终选择90%乙腈水(含2.0%甲酸)为提取液。
2.3 固相萃取柱的选择
该试验研究了3种通过式净化固相萃取小柱,即Oasis PRIME HLB、Captiva EMR-Lipid和Anavo HMR-Lipid。Oasis PRIME HLB可以有效吸附非极性干扰物,例如食品中的脂肪和磷脂,不需要活化和平衡步骤,直接可用于后续处理。Captiva EMR-Lipid和Anavo HMR-Lipid都能高效吸附脂类等杂质,不需要活化和平衡步骤,直接可用于后续处理。通过对3种固相萃取小柱的净化效果进行考察,结果发现(图3),Anavo HMR-Lipid对喹诺酮类药物的吸附性较强,不适合试验需求;Oasis PRIME HLB和Captiva EMR-Lipid的回收率均能满足要求。考虑到以90%乙腈水(含2.0%甲酸)为提取溶液时,Oasis PRIME HLB的过柱速度更快,且净化效果更好,因此选择Oasis PRIME HLB小柱。