红茶菌发酵液宏基因组分析
作者: 赵平 刘庆静 杨光 郭莹莹 邹积宏
摘要 对3个发酵批次的红茶菌发酵液进行宏基因组序列分析,采用数据库进行比对,发现红茶菌发酵液中存在短乳杆菌、植物乳杆菌、食窦魏斯氏菌、发酵乳杆菌、戊糖乳杆菌、融合魏斯氏菌、罗伊氏乳杆菌、柠檬明串珠菌、类植物乳杆、副干酪乳杆菌、乳酸片球菌等具有益生功能的细菌。宏基因组注释到的功能基因共有76 512个,其中与代谢过程注释到的功能基因数最多,且这些代谢途径上注释得到的菌株多数是益生菌株,主要参与了与碳水化合物代谢相关的三羧酸循环、柠檬酸循环、磷酸戊糖途径、糖酵解等;与氨基酸代谢相关主要有丙氨酸、天冬氨酸和谷氨酸代谢途径、甘氨酸、丝氨酸和苏氨酸等代谢途径。
关键词 红茶菌;益生菌;宏基因组;分析
中图分类号 TS272.7 文献标识码 A 文章编号 0517-6611(2024)09-0071-08
doi:10.3969/j.issn.0517-6611.2024.09.016
开放科学(资源服务)标识码(OSID):
Metagenomic Analysis of Kombucha Fermentation Broth
ZHAO Ping1,LIU Qing-jing2, YANG Guang3 et al
(1.Heilongjiang Green Food Science Research Institute, Harbin, Heilongjiang 150028;2.Heilongjiang University, Harbin, Heilongjiang 150008;3.Quality Supervision and Testing Institute of Heilongjiang Province, Harbin, Heilongjiang 150028)
Abstract The metagenomic sequence analysis of the fermentation broth of three fermentation batches of kombucha was carried out, and the database was used for comparison. It was found that there were mainly Lactobacillus brevis, Lactobacillus plantarum, Weisseria sinusosus, Lactobacillus fermentatum, Lactobacillus pentosus, Weisseria fused, Lactobacillus reuteri, Nebulococci citronicum, Lactobacillus paracasei, Lococci lactate and other bacteria with probiotic function in the fermentation broth of tea fungus. A total of 76 512 functional genes were annotated by metagenomes, among which the most functional genes were annotated by metabolic processes. Most of the strains annotated by these metabolic pathways were probiotic strains, which were mainly involved in the tricarboxylic acid cycle, citric acid cycle, pentose phosphate pathway, glycolysis and so on related to carbohydrate metabolism. The metabolic pathways of alanine, aspartate and glutamate, glycine, serine and threonine are mainly related to amino acid metabolism.
Key words Kombucha;Probiotics;Metagenome;Analysis
基金项目 国家市场监督管理总局技术保障专项项目(2020YJ010);黑龙江省财政支持项目。
作者简介 赵平(1985—),男,黑龙江哈尔滨人,副研究员,从事生物产品开发、食品微生物检测研究。
*通信作者,教授,从事食品和药物物质功能的挖掘及开发研究。
收稿日期 2023-06-02;修回日期 2023-07-08
红茶菌是民间用茶糖水与微生物培育菌苔或菌液而成,因多用红茶,菌液呈红色而得名。由于自然发酵过程而产生对人体有益的活性物质,不仅留有红茶本身的饮用价值,而且还具有独特的保健作用,曾风靡一时作为饮品流行至今[1]。红茶菌液发酵一定时间后,会形成苔状似海蜇皮,又称“海宝”[2]、“红茶菇”等[3-4]。红茶菌最早发现于约公元前220年,因具有解毒提神、助消化、抑菌、抗氧化、护肝、防癌、抗癌、降血糖、降血脂、调节血压、分解有毒物质等药用价值被视为珍宝,称为“胃宝”[5-24]。
红茶菌的菌群结构、发酵产物和化学成分随着现代分子生物学技术的快速发展而被逐步深入研究。目前研究表明,红茶菌是由多年来人工培养驯化多种共生益生菌组成,其中包括细菌和真菌[25]。在我国红茶菌虽然有很长的饮用历史,但仍停留在作坊式阶段,易染杂菌、风味变化大等问题导致无市场竞争力[26]。因此,探索红茶菌发酵液中微生物的群落结构,从基因层面解释微生物与发酵液风味及安全性之间的联系十分重要。近年来,宏基因组(Metagenome)测序广泛应用于奶酪、普洱茶、可可豆、发酵香肠、酒、酱油、食醋等发酵食品,且取得了一系列的研究成果,这对研究菌群结构及挖掘其功能基因至关重要[27]。
笔者利用宏基因组学,对红茶菌发酵液中微生物进行测序,对结果序列进行优化、组装、去冗余后注释物种,对微生物的群落结构、功能基因、代谢通路进行探索,从基因层面探讨微生物与红茶菌发酵液中的碳水化合物、氨基酸等代谢通路,阐明微生物在风味物质产生与形成过程中的作用。
1 材料与方法
1.1 主要材料
红茶菌,黑龙江质量监督检测研究院微生物与分子生物学检测研究中心保藏。
1.2 主要仪器和设备
AOEA580型HiSeq 2 500 高通量测序系统,美国 Illumina 公司;2266型洁净工作台,上海拜艾斯净化设备有限公司。
1.3 试验方法
1.3.1 样品检测。
分别取3批次实验室发酵7 d所得的红茶菌发酵液,进行宏基因组测序。
1.3.2 测序数据预处理及质量评估。
测序数据预处理后将符合要求的产物用 Illumina HiSeq 高通量测序平台进行测序。筛查下机的原始数据质量,去除非目的序列,获得高质量可用于宏基因组分析的数据集(Clean data),再对筛查出的Clean data拼接、校正与组装,构建宏基因组叠连群(Contigs)序列集,进行去冗余基因预测,获得非冗余氨基酸序列集,用于后续分析。
1.3.3 红茶菌微生物多样性分析。
物种α分析时首先对全体样本种水平的组成谱,在最低测序深度下进行随机重抽样,从而校正测序深度引起的多样性差异。随后,利用QIIME软件计算每个样本上述4种多样性指数(Chao1、ACE、Shannon、Simpson),对各样本在不同测序深度下的种水平丰度分布随机抽样,以各深度下抽取到的序列数与其对应的物种数绘制稀疏曲线。β多样性分析法,主要采用主成分分析法,通过QIIME和R软件对样本进行PCA分析,并描述样本间的差异分布特征。根据各样本在种水平的组成谱,使用R软件可计算共有、独有物种的数量,并通过Venn图直观地呈现各样本所共有和独有的物种数量。
1.3.4 红茶菌微生物功能基因分析。
1.3.4.1 红茶菌eggNOG数据库注释。
将预测得到的蛋白序列集与EggNOG数据库(http://eggnog.embl.de/version_5.0/,v5.0)的参考蛋白序列进行比对。比对软件使用MMseqs2,设置灵敏度参数为5.7,选择alignment得分最高的参考蛋白序列作为比对结果。通过eggnog-mapper将比对结果映射到EggNOG的数据库中,提取其分类信息及后续的GO注释结果。将注释得到的eggNOG结果及基因的丰度矩阵整合,可以获取各蛋白对应的eggNOG直系同源基因簇的丰度。再根据每个直系同源基因簇所属的分类就能统计不同水平的EggNOG功能类群丰度。
1.3.4.2 红茶菌KEGG代谢通路注释。
将预测得到的蛋白序列集与KOBAS软件的蛋白数据库(v2020_10_20)进行相似比对,比对软件使用MMseqs2,设置灵敏度参数为5.7,选择alignment得分最高的参考蛋白序列作为比对结果。KOBAS参考序列带有KO注释,通过将蛋白序列与之比对,即可获得蛋白序列的KO信息。将注释得到的KO结果及基因的丰度矩阵整合,可以获取各蛋白对应的KO丰度。最后再使用MinPath推断存在的代谢通路(KEGG pathway),并统计其丰度。
1.3.4.3 红茶菌GO功能注释。
蛋白编码基因的GO注释采用eggnog-mapper软件完成,得到的GO编号通过map2slim得到GOSlim注释结果,这一注释过程参考了记录GO信息的go-basic.obo文件(http://purl.obolibrary.org/obo/go/go-basic.obo)。将注释得到的GO结果及基因丰度矩阵整合,可以获取各蛋白对应的GO丰度。再根据每个GO所属的分类,可以统计不同水平的GO功能类群丰度。
1.3.4.4 红茶菌CAZy功能注释。
将预测得到的蛋白序列集与dbCAN(DataBase for automated Carbohydrate-active enzyme Annotation)中的参考蛋白序列(v2021_09_24)进行相似比对。比对软件使用MMseqs2,设置灵敏度参数为5.7,选择alignment得分最高的参考蛋白序列作为比对结果。将注释得到的CAZy功能类群结果及基因的丰度矩阵整合,可以获取各蛋白对应的CAZy酶家族的丰度。再依据酶所属的功能模块统计不同模块的CAZy酶家族丰度。
2 结果与分析
2.1 红茶菌微生物物种丰度及多样性
2.1.1 物种丰度。
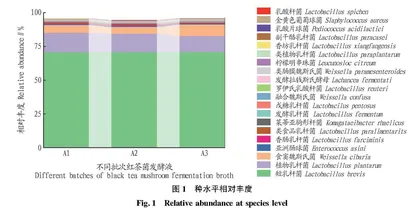
图1为选取种水平上的相对丰度柱状图,可直观反映出A1、A2、A3种水平上丰度排名前20的物种及比例。由图1可知,3个样品主要包括短乳杆菌、植物乳杆菌、食窦魏斯氏菌、亚洲肠球菌、香肠乳杆菌等。其中,短乳杆菌丰度最高,占总菌数的70.46%,A1、A2、A3中短乳杆菌分别占70.48%、70.34%、70.57%,说明发酵不同批次的短乳杆菌丰度差异较小,也再次证明了红茶菌发酵工艺的稳定性。红茶菌发酵液中还包含植物乳杆菌、食窦魏斯氏菌、亚洲肠球菌,分别占总菌数的13.34%、3.19%、0.65%,除此之外还有香肠乳杆菌、类食品乳杆菌、莱蒂亚驹形杆菌等。表1为对应图1整理出的相对丰度前十并被世界公认的益生菌。益生菌是一类对人体有益,可在肠道定殖并在肠道具有益生功能的一类微生物。由表1可知,红茶菌中的益生菌主要集中在短乳杆菌、植物乳杆菌、类植物乳杆菌、发酵乳杆菌,在人体中主要起抑菌、降胆固醇、调节机体免疫、维持肠道菌群等作用;食窦魏斯氏菌、副干酪乳杆菌、乳酸片球菌主要起抗氧化作用;戊糖乳杆菌可降血脂血糖、降尿酸,罗伊氏乳杆菌也可防溃疡、缓解过敏等症状。