高效液相色谱法测定保健食品中七烯甲萘醌的含量
作者: 张霞
维生素K2是一种脂溶性维生素,天然存在于动植物体内,是结构相似的一系列物质的总称。其中,七烯甲萘醌(MK-7)是指侧链含有七个双键的维生素K2类化合物,也是活性最高、功能最强的维生素K2化合物。MK-7在人体内参与骨代谢,具有预防骨质疏松的功效,还能诱导肝癌、胃癌、肺癌、骨肉瘤等癌细胞凋亡,具有较好的抗癌功效。市场监督管理总局于2016年发布的205号公告将MK-7收录于保健食品原料目录,批准采用发酵法生产的MK-7可作为保健食品的维生素K来源,适用于所有人群,每日允许摄入量为15—100μg。当前市售的维生素K2保健食品较多,但食品安全国家标准检验方法体系框架内暂时没有保健食品维生素K2的配套方法。市售保健食品中以添加反式结构MK-7为主,本实验旨在找到一种准确、稳定、高精密度、操作简单的高效液相色谱法,用于检测保健食品中的反式MK-7含量。
1. 仪器与材料
1.1 仪器
高效液相色谱仪,配荧光检测器(FLD)/二极管阵列检测器(DAD),U3000,美国赛默飞世尔;数控超声波清洗器,KQ2200DETB,昆山市超声仪器有限公司;涡旋仪,Vortex-Genie2,美国Scientific Industries公司;离心机,SF-TDL-4A,上海菲恰尔;恒温振荡箱,THZ-98AB,上海一恒科学仪器有限公司;色谱柱:SB-C18色谱柱,250mm*4.6mm,5μm,880975-902,美国安捷伦;锌粉还原柱:50mm*4.6mm,LAAA-0002-01,上海安谱实验科技股份有限公司。
1.2 材料
七烯甲萘醌,MK-7,含量:99.5%,USP挪威;异丙醇,色谱纯,北京迪马科技;甲醇,色谱纯,北京迪马科技;四氢呋喃,色谱纯,北京迪马科技;冰醋酸,分析纯,上海凌峰;无水乙酸钠,分析纯,上海凌峰;氯化锌,分析纯,上海凌峰;无水乙醇,分析纯,上海凌峰;碳酸钾,分析纯,上海凌峰;实验用水为超纯水。
2. 实验与方法
2.1 七烯甲萘醌(MK-7)标准储备液配制
准确称取MK-7标准品(CAS No:2124-57-4)10mg,用异丙醇溶解并转移至100mL棕色容量瓶中,再用异丙醇定容至刻度。标准储备液浓度为100μg/mL。
2.2 样品前处理
固体片剂样品均匀研磨成粉状,胶囊样品取内容物混合均匀,液体口服液样品混合均匀。精密称取1g样品至10mL棕色容量瓶中(用变性淀粉或糊精包埋过的样品先加0.01mol/L盐酸溶液1.0mL,涡旋混匀后超声10min破包埋;含油脂的软胶囊样品加脂肪酶0.2g,加水2.0mL,涡旋混匀后于37℃恒温振荡箱酶解4h以上),加异丙醇约5mL,涡旋混合至样品分散,超声提取30min,中间超声处理10min后,取出手动摇匀1次。超声完成后,取出放至室温,用异丙醇定容至刻度,摇匀,离心,取上清液过膜上机。
2.3 上机测试色谱条件
采用Agilent Zorbax SB-C18色谱柱(250mm*4.6mm,5μm,880975-902),上海安谱生产的锌粉还原柱(50mm*4.6mm,LAAA-0002-01)。流动相配制:甲醇900mL、四氢呋喃100mL、冰醋酸0.3mL、氯化锌1.5g和无水乙酸钠0.5g混合均匀,超声脱气30min备用。流动相上机流速为1.0mL/min,等度洗脱。采用FLD检测,激发波长(EX)为243nm,发射波长(EM)为430nm,色谱柱柱温为30℃,进样量10为μL。
2.4 标准曲线的绘制
精密移取MK-7标准储备液0.5mL至100mL棕色容量瓶中,用异丙醇溶解并定容至刻度,摇匀,此中间液浓度为0.5μg/mL。分别精密移取0.00、0.50、1.00、2.00、5.00mL中间液至10mL棕色容量瓶中,用异丙醇溶解并定容至刻度,摇匀,取中间液作为曲线最高点。MK-7标准曲线系列溶液的浓度分别为0.000、0.025、0.050、0.100、0.250、0.500μg/mL,各取10μL,按照上述色谱条件上机测试,记录色谱图峰面积,以标准溶液浓度为横坐标、峰面积为纵坐标绘制标准曲线。
3. 结果与分析
3.1 实验条件优化
3.1.1 前处理优化。原卫计委《关于海藻酸钙等食品添加剂新品种的公告(2016年第8号)》附件3方法中的测试基质为维生素K2(发酵法)油剂产品与粉剂产品,此类基质为生产MK-7保健食品的原料。本研究参照原卫计委方法中的样品前处理方式,采用异丙醇超声提取30min后过膜上机,色谱峰分离度较好,峰形对称良好,MK-7测试值能达到样品理论添加量的90%以上,复现性较好,偏差不超过10%。同时,本实验前处理还参考了GB 5009.290-2023《食品安全国家标准 食品中维生素K2的测定》中的提取方式,样品经脂肪酶、淀粉酶在37℃恒温箱酶解2h以上,加入10mL无水乙醇及1.0g碳酸钾,混匀后分2次(每次10mL)加入正己烷20mL,振荡提取10min,再离心分层,合并正己烷层至100mL旋蒸瓶中,旋蒸至干,用流动相复溶,混匀后过膜上机,色谱峰分离度较好,峰形对称良好,但MK-7测试值偏低,不到理论添加量的80%,且复现性较差,偏差在30%以上。因此,前处理确定采用异丙醇超声提取的方式,操作简单,测试结果符合理论预期,复现性较好。
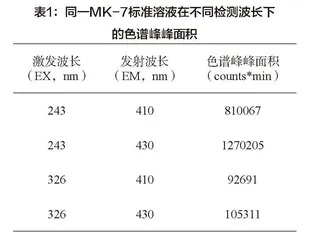
3.1.2 高效液相色谱上机条件优化。本实验对比了原卫计委《关于海藻酸钙等食品添加剂新品种的公告(2016年第8号)》附件3、GB 5009.290-2023《食品安全国家标准 食品中维生素K2的测定》、GB 5009.158-2016《食品安全国家标准 食品中维生素K1的测定》第一法的上机条件。第一种方法的上机测试条件是:采用Agilent Zorbax SB-C18色谱柱,流动相为纯甲醇,流速为1.0mL/min,等度洗脱,DAD检测,波长为254nm,柱温为50℃,进样量为20μL。通过验证,此方法的定量限为10μg/g。目前实验室检测过的各种保健食品中MK-7含量为0.6-360μg/g,因此原卫计委方法中的上机条件不适用于低含量保健食品的检测。维生素K1、维生素K2在国标方法中的上机测试条件是:色谱柱、流动相、流速、柱温、进样量参照上述色谱条件,采用FLD检测,维生素K1的EX为243nm、EM为430nm,维生素K2的EX为326nm、EM为410nm。两种方法除了检测波长不一样,其余色谱条件都一样。表1统计对比了同一浓度的MK-7标准溶液在不同波长下检测到的色谱峰峰面积,由此可知,在EX为243nm、EM为430nm检测波长下,MK-7响应最好。
3.2 方法线性范围及定量限
MK-7标准曲线回归方程为Y=1040879X(R2=0.9999),MK-7在0.00—0.50μg/mL与色谱峰峰面积呈正相关。称取1.0g维生素K口服液空白样品至10mL棕色容量瓶中,共10份,每份添加0.25μg的MK-7,进行样品处理后上机测试,计算样品中的MK-7含量。计算10个空白样品加标含量的标准偏差SD,分析方法的检出限(LOD)为3SD对应的待分析物含量,定量限(LOQ)为10SD对应的待分析物含量。结果显示,方法检出限为0.0408μg/g,定量限为0.136μg/g。根据实验室测试经验,大部分保健食品的MK-7含量在0.6—360μg/g,故此方法定量限能够满足同类产品的定量测试需求。
3.3 精密度与耐用性
称取6份维生素K口服液阳性样品、6份维生素K咀嚼片阳性样品各1.0g至10mL棕色容量瓶中,进行样品处理后,检测样品的MK-7含量,计算其相对标准偏差RSD值,口服液为2.18%,咀嚼片为1.21%,详见表2。取1号、2号口服液样品溶液进行0.9、1.0、1.1mL/min不同流速上机测试的耐用性试验,计算其RSD值为3.85%;取1号、2号口服液样品溶液进行28、30、32℃不同柱温上机测试的耐用性试验,计算其RSD值为3.01%;取1号口服液样品溶液进行0、8、16、24h不同存放时间后的上机测试耐用性试验,计算其RSD值为1.24%。以上数据说明,本分析方法具有良好的精密度与耐用性。
3.4 准确度
称取维生素K口服液空白样品9份各1.0g至10mL棕色容量瓶中,平均分成3组,每组3个样品,分别向每组添加0.5、1.0、1.5μg的MK-7标准物质,进行样品处理后,检测样品中MK-7加标回收率。结果显示,回收率范围为94.4%—99.6%,RSD值为1.82%,说明本方法的准确度较好。
综上所述,本方法准确度高、精密度高、耐用性好,能够满足绝大部分市售保健食品中七烯甲萘醌含量的检测需求。不过,目前本方法只能检测CAS号为2124-57-4的反式结构MK-7,对于其异构体(CAS:27670-94-6)则会在目标位出现五指峰。用变性淀粉或糊精包埋处理的样品需要添加盐酸溶液破包埋,含油量高的样品则需要用脂肪酶酶解后再进行下一步操作。同时,因为MK-7对光敏感,所以整个操作过程必须严格避光。
作者简介:张霞(1983—),女,汉族,重庆人,工程师,大学本科,研究方向为食品中维生素含量的检测。