超高效液相色谱-质谱联用法测定猪肉中4种喹诺酮类药物残留
作者: 陈美娜
摘 要:建立超高效液相色谱-质谱联用法测定猪肉中恩诺沙星、环丙沙星、氧氟沙星、沙拉沙星4种喹诺酮类药物残留的分析方法。试样经EDTA-Mcllvaine缓冲溶液提取,用HLB固相萃取柱净化,氮吹浓缩后用1 mL甲酸水溶液(0.1%)复溶,上机测定。结果表明,4种喹诺酮类药物残留组分在2.5~100.0 μg·L-1线性关系良好,方法检出限为1.0~2.5 μg·kg-1,定量限为3.0~8.0 μg·kg-1,加标回收率在90.0%~109.2%,相对标准偏差(Relative Standard Deviation,RSD)为1.01%~5.74%,适用于猪肉中4种喹诺酮类药物残留的检测。
关键词:超高效液相色谱-质谱联用法;猪肉;喹诺酮
Determination of Four Quinolone Residues in Pork by Ultra-High Performance Liquid Chromatography-Mass Spectrometry
CHEN Meina
(Shenzhen Academy of Metrology & Quality Inspection, Shenzhen 518000, China)
Abstract: Establish an analytical method for the determination of residues of four quinolone drugs, including enrofloxacin, ciprofloxacin, ofloxacin, and sarafloxacin, in pork using ultra-high performance liquid chromatography-mass spectrometry. The sample was extracted with EDTA Mcllvaine buffer solution, purified with HLB solid-phase extraction column, concentrated with nitrogen blowing, and then dissolved in 1 mL formic acid aqueous solution (0.1%) for measurement on the machine. The results showed that there was a good linear relationship between the residual components of the four quinolone drugs in the range of 2.5~100.0 μg·L-1. The detection limit of the method was 1.0~2.5 μg·kg-1, and the quantification limit was 3.0~8.0 μg·kg-1. The recovery rate was in the range of 90.0%~109.2%, and the relative standard deviation (RSD) was 1.01%~5.74%. It is suitable for the detection of four quinolone drug residues in pork.
Keywords: ultra high performance liquid chromatography-mass spectrometry; pork; quinolone
喹诺酮类药物是由人工合成的一类人畜通用的抗菌药物,其凭借高效低毒、抗菌谱广、无交叉耐药性等优点而广泛用于动物疾病治疗[1-2]。然而,动物、水产养殖过程中过量或超范围使用该类药物,通过食物链进入人体后容易产生耐药菌株,且有潜在的致癌和遗传毒性[3-4]。猪肉是公众最常食用的肉类之一,加强猪肉中的兽药残留监管对公众的身体健康非常重要。本研究参照《动物源性食品中14种喹诺酮药物残留检测方法 液相色谱-质谱/质谱法》(GB/T 21312—2007)[5],建立了超高效液相色谱-质谱联用法(Ultra High Performance Liquid Chromatography-Mass Spectrometry,UPLC-MS)测定猪肉中4种喹诺酮类药物残留的方法。
1 材料与方法
1.1 材料与试剂
猪肉,来源于深圳市辖区内抽检样品。
恩诺沙星标准溶液(CDAA-S-550003-AD-1 mL)、环丙沙星标准溶液(CDAA-S-550001-AD-1.2 mL)、氧氟沙星标准溶液(CDAA-S-550006-AD-1.2 mL)、沙拉沙星标准溶液(CDAA-S-530219-AD-1.2 mL),浓度均为1 000 μg·mL-1,由上海安谱璀世标准技术服务有限公司提供;甲醇,德国默克公司提供;甲酸(99%),山东旭晨化工科技有限公司。
Mcllvaine缓冲溶液:将1 000 mL柠檬酸(0.1 mol·L-1)与625 mL磷酸氢二钠(0.2 mol·L-1)混合,调节缓冲溶液pH值为4.0±0.05。
EDTA-Mcllvaine缓冲溶液:称取60.5 g乙二胺四乙酸二钠,加入1 625 mL Mcllvaine缓冲溶液,振荡摇匀。
1.2 仪器与耗材
TSQ Quantis高效液相色谱-质谱联用仪,美国赛默飞公司;GL2202-1SCN分析天平(精度0.01 g),德国赛多利斯公司;HT190R高速台式冷冻离心机,湖南湘仪实验室仪器开发有限公司;PHS-3E pH计,上海仪电科学仪器股份有限公司;ASE-12固相萃取装置,天津奥特赛恩斯仪器有限公司;SelectCore HLB固相萃取柱,纳谱分析技术(苏州)有限公司。
1.3 试验方法
1.3.1 样品前处理
准确称取5.00 g(精确至0.01 g)均质过的猪肉试样于50 mL螺口离心管中,加入20 mL EDTA-Mcllvaine缓冲溶液(0.1 mol·L-1),用涡旋混合仪涡旋混合1 min,置于超声波清洗器中超声10 min,再于4 ℃、10 000 r·min-1的离心机中离心5 min,反复提取3次,合并上清液于试管中。
将HLB固相萃取柱置于固相萃取装置上,先用6 mL甲醇冲洗,再用6 mL超纯水活化,然后将提取得到的上清液倒入固相萃取小柱,控制以3 mL·min-1的速度过柱,弃去过滤液,再用2 mL甲醇水溶液(5%)淋洗,弃去淋洗液,抽干萃取小柱,用6 mL甲醇洗脱,收集洗脱液于试管中,置于氮吹仪中通入氮气直至吹干,加入1 mL甲酸水溶液(0.1%)溶解,涡旋混合,转移入进样小瓶中上机测定。
1.3.2 标准系列溶液配制
分别吸取1.0 mL浓度均为1 000 μg·mL-1的恩诺沙星、环丙沙星、氧氟沙星、沙拉沙星标准溶液于100 mL容量瓶中,用甲醇稀释并定容至刻度,配制成4种喹诺酮类药物组分浓度均为10 μg·mL-1的混合标准储备液。
将混合标准储备液用甲酸水溶液(0.1%)逐渐稀释成4种喹诺酮类药物组分浓度分别为2.5 μg·L-1、5.0 μg·L-1、10.0 μg·L-1、20.0 μg·L-1、40.0 μg·L-1、80.0 μg·L-1、100.0 μg·L-1的混合标准工作系列。称取5.0 g阴性基质样品,依次加入1.0 mL混合标准工作系列溶液,按1.3中试样处理过程提取并净化。
1.3.3 仪器分析条件
(1)液相色谱条件。色谱柱:ZORBAX SB-C18(100 mm×2.1 mm,1.8 μm);柱温:40 ℃;流速:0.3 mL·min-1;进样量:2 μL;流动相:A甲酸水溶液(0.1%)、B甲醇;梯度洗脱程序:0~0.2 min,10%B;0.2~2.5 min,10%B→50%B;2.5~3.2 min,50%B→85%B;3.2~4.5 min,85%B保持1min;4.5~5.0 min,85%B→10%B;5.0~6.0 min,10%B。
(2)扫描方式:选择反应监测(Selected Reaction Monitoring,SRM)模式;毛细管电压:3 500 V;离子源温度:300 ℃;鞘气压力:55 bar;监测离子:恩诺沙星360.3>316.4(定量),360.3>342.3(定性);环丙沙星332.2>314.3(定量),332.2>288.3(定性);氧氟沙星362.2>318.3(定量),362.2>261.2(定性);沙拉沙星386.3>342.3(定量),386.3>299.3(定性)。
2 结果与分析
2.1 线性方程与相关系数
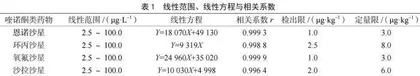
以混合标准工作系列中恩诺沙星、环丙沙星、氧氟沙星、沙拉沙星的浓度(X)为横坐标,相对应测得的定量离子峰面积(Y)为纵坐标,由仪器工作站自动绘制标准工作曲线,计算线性回归方程与相关系数,见表1。由表1中数据可知,4种喹诺酮类药物组分在2.5~100.0 μg·L-1线性关系良好,相关系数r均大于0.99,满足相关标准对线性的要求。
2.2 方法检出限与定量限
称取5.00 g经检测不含恩诺沙星、环丙沙星、氧氟沙星、沙拉沙星的阴性猪肉试样,添加适量4种喹诺酮类药物组分混合标准中间液进行测试,以4种喹诺酮类药物组分的定性离子的重构离子色谱峰的信噪比S/N≥3对应浓度来确定方法检出限,以定量离子的重构离子色谱峰的信噪比S/N≥10对应浓度来确定方法定量限,见表1。由表1中数据可知,4种喹诺酮类药物组分的方法检出限为1.0~2.5 μg·kg-1,方法定量限为3.0~8.0 μg·kg-1,检出限与定量限均符合方法标准《动物源性食品中14种喹诺酮药物残留检测方法 液相色谱-质谱/质谱法》(GB/T 21312—2007)中的规定。
2.3 加标回收率及精密度
称取18份经检测不含上述4种喹诺酮类药物组分的阴性猪肉试样,每6份为一组,分别添加低(2.5 μg·kg-1)、中(20.0 μg·kg-1)、高(80.0 μg·kg-1)3个浓度水平的4种喹诺酮类药物混合标准溶液,按照1.3.1样品前处理过程进行提取、净化,1.3.3仪器分析条件进行测试,计算加标回收率与精密度(以相对标准偏差表示),见表2。由表2数据可知,恩诺沙星加标回收率为99.1%~108.8%,相对标准偏差(Relative Standard Deviation, RSD)为1.01%~2.95%;环丙沙星加标回收率为90.0%~109.2%,RSD为3.07%~5.74%;氧氟沙星加标回收率为97.7%~109.1%,RSD为1.99%~2.59%;沙拉沙星加标回收率为95.6%~106.7%,RSD为1.18%~2.73%,回收率与精密度均符合《动物源性食品中14种喹诺酮药物残留检测方法 液相色谱-质谱/质谱法》(GB/T 21312—2007)中关于回收率和RSD的要求。
3 结论
本研究参照GB/T 21312—2007,建立了UPLC-MS法测定猪肉中4种喹诺酮类药物残留的方法,恩诺沙星、环丙沙星、氧氟沙星、沙拉沙星等组分在2.5~100.0 μg·L-1线性关系良好,方法检出限为1.0~2.5 μg·kg-1,定量限为3.0~8.0 μg·kg-1,加标回收率在90.0%~109.2%,RSD为1.01%~5.74%,可用于检验检测机构对猪肉中4种喹诺酮类药物残留的抽检检测。
参考文献
[1]龙启萍,曹海兰,吕东晋,等.超高效液相色谱-质谱串联法测定牦牛肉中11种喹诺酮类药物残留[J].医学动物防制,2018,34(6):570-573.
[2]孙晓,王涛,王永姣,等.超高效液相色谱-质谱串联法测定蜂蜜中喹诺酮类药物残留[J].食品安全质量检测学报,2019,10(17):5604-5608.
[3]张威,胡婷婷,秦培余,等.液相色谱-质谱/质谱法测定鸡蛋粉中喹诺酮类药物残留[J].现代食品,2022,28(20):155-158.
[4]袁荷芳,耿成钢,高蕙文,等.超高效液相色谱-串联质谱法测定水产品中16种喹诺酮类药物残留[J].中国食品,2021(19):66-67.
[5]中华人民共和国国家质量监督检验检疫总局,中国国家标准化管理委员会.动物源性食品中14种喹诺酮药物残留检测方法 液相色谱-质谱/质谱法:GB/T 21312—2007[S].北京:中国标准出版社,2007.