超高效液相串联质谱法测定牛奶中8种氟喹诺酮类兽药残留
作者: 张瑞婷
摘 要:建立了超高效液相串联质谱法同时测定牛奶中8种氟喹诺酮类兽药残留的分析方法。牛奶样品用提取剂提取后,用固相萃取柱净化,选用C18色谱柱梯度洗脱分离。通过串联质谱在电喷雾电离-正离子、多反应监测模式下进行定性和定量分析。结果表明,8种兽药在0.5~500.0 μg·mL-1呈良好的线性关系,R2>0.996,在10 μg·kg-1、50 μg·kg-1、100 μg·kg-1 3个浓度添加水平下,回收率为81.3%~98.8%,RSD为2.3%~9.8%。该方法灵敏、快速、可靠,适用于牛奶中多种兽药残留的快速筛查。
关键词:超高效液相串联质谱法;氟喹诺酮类;牛奶
Determination of 8 Fluoroquinolones Veterinary Drug Residues in Milk by Ultra Performance Liquid Chromatography Tandem Mass Spectrometry
ZHANG Ruiting
(Beijing Institute of Product Quality Supervision and Inspection, Beijing 101300, China)
Abstract: A method for the simultaneous determination of eight fluoroquinolone veterinary drug residues in milk by ultra performance liquid chromatography tandem mass spectrometry was established. After the milk sample was extracted with the extractant, it was purified by a solid phase extraction column and separated by gradient elution on a C18 chromatographic column. Qualitative and quantitative analyzes were then performed by tandem mass spectrometry in electrospray ionization-positive ion, multiple reaction monitoring mode. The results showed that the eight veterinary drugs showed a good linear relationship in the range of 0.5~500.0 μg·mL-1, R2>0.996. The recoveries ranged from 81.3% to 98.8% at the spiked levels of 10 μg·kg-1, 50 μg·kg-1, 100 μg·kg-1 with the RSD of 2.3% to 9.8%. The method is sensitive, rapid and reliable, and is suitable for rapid screening of various veterinary drug residues in milk.
Keywords: ultra performance liquid chromatography tandem mass spectrometry; fluoroquinolones; milk
氟喹诺酮类药物为白色或淡黄色晶型粉末,属于广谱抗生素,常被用于预防和治疗奶牛疾病[1]。兽药的过量使用或不当使用与公众的健康息息相关,长期、低水平的接触方式不仅导致细菌产生耐药性,还会产生各种慢性、蓄积性毒害,给消费者带来潜在危险。因此,建立兽药残留的分析方法对于风险监控是十分必要的[2]。目前,兽药残留量的检测方法主要有金标检测法、酶联免疫吸附法、高效液相色谱法、气相色谱法和质谱法等[3]。超高效液相串联质谱法的优点是选择性强、应用范围广、分离效果好、灵敏度高和能自动化操作[4]。其可以把待测组分分离与结构的定量分析有机结合在一起,特别适用于药物残留量和药物的确证分析[5]。
1 材料与方法
1.1 仪器与试剂
超高效液相串联质谱仪(Waters公司);氮吹浓缩仪(Organomatian Associates公司);冷冻离心机(Sigma公司);Oasis HLB固相萃取柱(200 mg,6 mL,Waters公司)。
8种喹诺酮类兽药标品:恩诺沙星、诺氟沙星、环丙沙星、司帕沙星、氧氟沙星、奥比沙星、萘啶酸和噁喹酸,均购于德国DR公司;乙腈、甲醇、正己烷和甲酸(HPLC级);超纯水(Mili-Q公司)。
1.2 溶液的配制
1.2.1 标准储备溶液
精密称量各种药品标准品10 mg(精确到0.1 mg)于10 mL容量瓶(棕色)中,用乙腈超声溶解并定容至刻度(其中噁喹酸需加少量NaOH溶液溶解),配制成1.0 mg·mL-1标准储备溶液,4 ℃冰箱避光保存。
1.2.2 混标工作溶液
取洗净烘干的50 mL棕色容量瓶,加入500 μL储备液,乙腈定容至刻度,配制成浓度为10.0 μg·mL-1的混标中间溶液,置于4 ℃冰箱冷藏避光保存。
1.2.3 流动相
0.3%甲酸水溶液:准确吸取3 mL甲酸至1 000 mL容量瓶,用超纯水定容至刻度,现配现用。流动相:乙腈∶0.3%甲酸=10∶90。
1.3 样品前处理
1.3.1 提取
将牛奶样品轻晃混匀后称取5.00 g(精确到0.01 g),装入50 mL的PP管中,依次加入无水硫酸钠5 g,1%的甲酸-乙腈溶液20 mL,轻柔涡旋混匀,充分反应。超声波提取时,保持处于冰浴环境中,离心转速5 000 r·min-1,时间5 min,之后取上清液于另一洁净的离心管中。将剩余残渣重复一次上述操作,取上清液。吸取10 mL提取到的上清液,加入20 mL乙腈饱和的正己烷,振荡静置分层弃去正己烷层,氮气吹至近干。
1.3.2 净化
HLB固相萃取柱(200 mg,6 mL)使用前用6 mL甲醇洗涤、6 mL水净化,将提取的上清液以2~3 mL·min-1的速度过柱,过两次后弃去滤液,用2 mL 5%的甲醇溶液淋洗,弃去淋洗液,减压真空泵抽取5 min,在吸附柱中加入5 mL甲醇洗脱液,5 000 r·min-1离心1 min,将收集到的洗脱液在氮气下吹至近干后,加入1 mL流动相溶液溶解,经0.22 μm微孔滤膜过滤,以备上机分析测定使用。
1.4 测定条件
1.4.1 色谱条件
色谱柱:Acquity UPLC C18色谱柱(2.1 mm×50 mm,1.7 µm BEH颗粒,美国沃特世公司);流动相:A相为乙腈,B相为0.3%甲酸水溶液;流速:0.3 mL·min-1;柱温:30 ℃;进样体积:5 μL。
1.4.2 质谱条件
离子源:电喷雾电离-正离子模式(ESI+);检测方式:MRM(多反应监测);毛细管电压:3.5 kV;脱溶剂气流量:800 L·h-1;锥孔气流量:120 L·h-1;碰撞气:氩气;碰撞气压:0.18 Pa。
2 结果与分析
2.1 提取剂的选择
在酸性提取溶剂中,氟喹诺酮类药物更容易被萃取;乙腈属于极性溶剂,可避免从样品中提取过多的脂肪,同时具有很好的蛋白沉积效果,可沉积样品中99%的蛋白,避免其他杂质的干扰。因此,初步确定采用乙腈为提取溶剂。
本实验对纯乙腈、1%乙酸-乙腈、1%甲酸-乙腈3种提取剂提取效果进行了对比,结果显示酸化乙腈提取效果更佳。其中1%甲酸-乙腈、1%乙酸-乙腈的提取效果相差不大,但1%甲酸-乙腈提取后的色谱峰更加尖锐,故确定最终提取溶剂为1%甲酸-乙腈。
2.2 流动相的选择及质谱条件的优化
以不同比例的乙腈-水为流动相进行梯度洗脱时,为了提高正离子化效率并改善峰形,同时考虑到分析组分中大多含有氨基和羧基基团,所以分别尝试了在水相中加入0.1%甲酸、0.2%甲酸、0.3%甲酸、5 mmol·L-1甲酸铵、10 mmol·L-1甲酸铵进行洗脱。对比结果显示,流动相中加入0.3%甲酸时,样品由结合态变为游离态,灵敏度高,分离效果好。
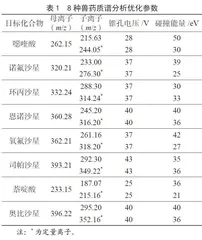
质谱参数的优化是通过ESI探针分别将8种兽药(浓度1 mg·L-1)的混合标准品工作液在正电离模式下用质谱蠕动泵自动进样,对每一种兽药进行母离子全扫描确定各种待测兽药的分子离子,而后取其分子离子为母离子,对其进行全扫描。选取丰度最强、干扰最小的两个离子用于目标分子的定量和定性分析,表1为8种兽药质谱分析优化参数。
2.3 线性范围及检出限
将8种待测兽药配制成一系列标准工作溶液,绘制标准工作曲线,对目标物质进行定量分析。分别得到待测兽药的标准曲线回归方程、定量限及相关系数,结果表明在0.5~500.0 μg·mL-1,R2>0.996,8种兽药的回归方程均线性良好。通过逐级稀释,根据信噪比(S/N)等于10确定为定量下限,结果见表2。
2.4 回收率与精密度
在待测样品中添加8种兽药的混标工作溶液,分别在10 μg·kg-1、50 μg·kg-1、100 μg·kg-1 3个浓度水平下进行回收率实验,同时每个浓度水平下进行6次平行实验,计算精密度。结果见表3。结果显示,该方法的平均回收率为81.3%~98.8%,RSD为2.3%~9.8%,表明本方法的回收率和RSD均在标准范围内,符合多残留分析的要求。
2.5 实际样品检测
在市场上随机购买15份不同批次牛奶样品,经检测发现1份阳性样品。恩诺沙星检出值12.1 µg·kg-1,其他药物无检出。该例阳性样品兽药残留量未超过GB 31650—2019规定的最高残留限量,可认为是合格样品。
3 结论
本实验建立了一种简单、灵敏、快速的8种兽药残留分析方法,通过对净化方法、流动相、质谱条件等一系列实验条件的探索优化,建立了UPLC-MS/MS同时检测牛奶中8种氟喹诺酮类兽药的分析方法。该方法克服了检测种类少、检测时间长、实验成本高等问题,适用于牛奶样品多残留的快速筛查和检测。
参考文献
[1]梁莉,李皓晨.浅析兽药残留的原因、危害及防控措施[J].中国食品,2021(19):88-89.
[2]周迎春,韩文凤,刘少博.我国动物源性食品中兽药残留的原因分析[J].粮食与油脂,2021,34(5):160-162.
[3]刁艳艳.LC-MS/MS法同时测定牛奶样品中25种兽药残留[D].郑州:郑州大学,2017.
[4]董李学,张立田,项爱丽,等.Qu ECHERS-LC-MS/MS法测定牛奶中喹诺酮类药物[J].食品工业,2019,40(1):314-317.
[5]刘晋源,杨蕴嘉,姚凯,等.超高效液相色谱-串联质谱法同时测定黑巧克力中4种牛奶过敏原蛋白[J].卫生研究,2022,51(2):286-292.