高效液相色谱法测定蜂蜜中诺氟沙星残留
作者: 魏超田 高倩妮
摘 要:本文建立了一种快速、高效、重复性好的高效液相色谱法-荧光检测器检测蜂蜜中的诺氟沙星药物残留量的方法。对蜂蜜样品中诺氟沙星的提取过程进行了优化,确定了最佳前处理方案:先用0.02 mol·L-1氢氧化钠溶解蜂蜜将其制成稀释液,然后用0.5%乙酸的乙腈溶液提取蜂蜜中的诺氟沙星,水浴蒸至近干,最后用0.2%甲酸溶解样品的残渣。其中,当蜂蜜和
0.02 mol·L-1氢氧化钠溶液的比例为1∶1,蜂蜜和0.5%乙酸的乙腈溶液比例为1∶5时诺氟沙星提取量最大。通过考察流动相种类、比例及流速对诺氟沙星保留和分离的影响,得出流动相为0.2%甲酸溶液-乙腈,0.2%甲酸和乙腈的比例为87∶13,流速为1.5 mL·min-1,柱温35 ℃,荧光检测激发波长280 nm,发射波长450 nm。采用本方法进行检测时,诺氟沙星的检出限为0.4 μg·kg-1,在0.001~0.200 μg·mL-1呈良好的线性关系(R2=0.995 7),加标回收率为84.98%~105.72%,本试验方法能够满足残留分析的要求。
关键词:高效液相色谱;蜂蜜;诺氟沙星;残留
Determination of Norfloxacin Residue in Honey by HPLC
WEI Chaotian1,2, GAO Qianni1,3
(1.Anhui Zhengjian Inspection and Test Company Lminted, Bozhou 236800, China; 2.Anhui Wanhuacao Biotechnology Company Lminted, Bozhou 236800, China; 3.Bozhou University, Bozhou 236800, China)
Abstract: A rapid, efficient and reproducible method for the determination of norfloxacin residues in honey by high performance liquid chromatography with fluorescence detector was established. The extraction process of norfloxacin from honey samples was optimized, and the best pretreatment scheme was determined: firstly, the honey was dissolved with 0.02 mol·L-1 sodium hydroxide to make a diluent, then the norfloxacin in honey was extracted with 0.5% acetic acid acetonitrile solution, evaporated in a water bath to nearly dry, and finally the residue of the sample was dissolved with 0.2% formic acid. Among them, when the ratio of honey to 0.02 mol·L-1 sodium hydroxide solution is 1∶1, and the ratio of honey to 0.5% acetic acid acetonitrile solution is 1∶5, the extraction amount of norfloxacin is the largest. By investigating the effect of mobile phase type, proportion and flow rate on the retention and separation of norfloxacin, the mobile phase was 0.2% formic acid solution-acetonitrile, the ratio of 0.2% formic acid and acetonitrile was 87∶13, and the flow rate was 1.5 mL·min-1. The column temperature was 35 ℃, the fluorescence detection excitation wavelength was 280 nm, and the emission wavelength was 450 nm.
When this method was used for detection, the detection limit of norfloxacin was 0.4 μg·kg-1, with a good linear relationship between 0.001 μg·mL-1 and 0.200 μg·mL-1(R2=0.995 7), and the recovery rate of standard addition was
84.98 %~105.72%. This test method can meet the requirements of residue analysis.
Keywords: hig performance liquid chromatography; honey; norfloxacin; residue
随着人们对健康和天然保健产品的追求和崇尚,具有抗氧化性、抗菌性等生物活性功能的蜂蜜及其产品的质量受到了人们的广泛关注。蜂蜜在养殖过程中会感染疾病或受到虫害,峰农为控制病虫害的发生而滥用抗生素,导致农药残留问题的发生。诺氟沙星属喹诺酮类药物,是在喹诺酮母核的基础上进行人工合成的一类广谱抗菌药,因其价格低、抗菌性强,被广泛用于人和动物疾病的治疗[1]。但其自身无法分解,在人体内会大量积累残留,人们食用动物组织后可能出现对该药物的严重耐药性。诺氟沙星在蜂蜜养殖过程中属于禁用药物,一经检出即为不合格产品[2]。2019年国家市场监督管理总局对蜂蜜抽检项目中,新增了几项喹诺酮类,其中诺氟沙星残留就是其中一项[3]。
目前测定诺氟沙星残留量的方法有微生物法、高效液相色谱-紫外或二极管阵列检测[4-6]、薄层色谱法[7-9]以及高效液相色谱-串联质谱法(High Performance Liquid Chromatography tandem Mass Spectrometry,HPLC-MS/MS))[10-13]。其中质谱法运用较多,质谱法虽然检出限低但操作复杂、耗费时间长,且由于设备条件和成本的限制无法普及。相关学者对诺氟沙星药物残留的检测方法研究大多集中在畜产品、奶制品、水产品上[14-16],对于蜂蜜中的诺氟沙星残留检测较少,且动物性等产品的前处理方法不适用蜂蜜。本试验通过研究蜂蜜不同的前处理方式,建立了一种简单、经济、高效、实用性强的高效液相色谱法测定蜂蜜中的诺氟沙星,以期为快速检测蜂蜜中诺氟沙星含量奠定理论基础,为蜂产品质量控制提供相应保证。
1 材料与方法
1.1 材料与试剂
诺氟沙星标准样品;乙腈(色谱纯),甲酸、乙酸、氢氧化钠、无水硫酸镁、磷酸氢二钠以及磷酸氢二钾均为分析纯,国药基团;蜂蜜(5种不同品牌)购自超市。
1.2 仪器与设备
高效液相色谱仪(配有紫外、荧光检测器),赛默飞世尔科技(中国)有限公司;AcclaimTM PolarAdvantage Ⅱ C18柱(250 mm×4.6 mm,5 μm)。
1.3 试验方法
1.3.1 标准曲线的绘制
准确量取100 μg·mL-1的诺氟沙星标液10 μL,用0.2%甲酸水溶液定容至1 mL,配制成1.00 μg·mL-1标准储备液。准确移取储备液按梯度稀释配制成浓度为0.001 μg·mL-1、0.002 μg·mL-1、0.004 μg·mL-1、0.080 μg·mL-1、0.010 μg·mL-1和0.200 μg·mL-1的标准系列工作液,储备于冰箱中备用。
1.3.2 高效液相色谱条件
色谱柱:AcclaimTM PolarAdvantage Ⅱ C18柱(250 mm×4.6 mm,5 μm);荧光检测波长:激发波长280 nm,发射波长450 nm;柱温:30 ℃;流速:1.5 mL·min-1;进样量10 μL;流动相:流动相A(乙腈)∶流动相B(0.2%甲酸水溶液)为13∶87(体积比,下同)。
1.3.3 蜂蜜样品处理
(1)蜂蜜稀释液的选择。①先用水溶解样品,再用0.5%乙酸的乙腈溶液萃取蜂蜜中的诺氟沙星。②用0.02 mol·L-1氢氧化钠溶液溶解样品,再用0.5%乙酸的乙腈溶液萃取蜂蜜中的诺氟沙星。③用Mallvaine pH 4.0缓冲液溶解样品,再用0.5%乙酸的乙腈溶液提取,最后水浴蒸发浓缩提取诺氟沙星。
称取约5.00 g(精确到0.01)蜂蜜,加入5.0 mL 0.02 mol·L-1氢氧化钠溶液,搅拌均匀,加入25 mL 0.5%乙酸乙腈溶液,涡旋5 min,加入无水硫酸镁5 g,涡旋2 min,4 000 r·min-1离心5 min,取上清液。
固相萃取柱依次用甲醇、磷酸缓冲盐各2 mL预淋,取上清液过柱,10 mL甲醇洗脱,收集液水浴蒸至近干,0.2%乙酸水溶液溶解定容至2 mL,过0.45 μm有机滤膜,高效液相色谱仪分析测定。
(2)提取液的选择。选取0.05 mol·L-1磷酸缓冲盐溶液、0.5%乙酸的乙腈溶液、乙腈+水(8∶2)混合溶液为蜂蜜中诺氟沙星的提取液,考察不同提取液及其添加量对蜂蜜中诺氟沙星提取量的影响。
2 结果与分析
2.1 高效液相色谱方法的建立
2.1.1 流动相及其比例的确定
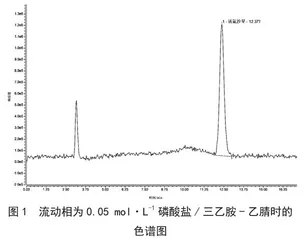
目前高效液相色谱法测定喹酮类含量时,一般选择磷酸/三乙胺、磷酸/庚烷磺酸钠、磷酸/四丁基溴化铵体系作为流动相分离喹诺酮类,但这几种体系中都含有盐类物质,易结晶,测定中容易造成色谱柱和检测器堵塞,极大缩短了色谱柱的使用寿命。
试验中发现荧光检测器对含有乙腈的流动相变化较敏感,且在流动相中加入适量的甲酸可以提高其灵敏度,因此本试验在乙腈-0.2%甲酸,0.05 mol·L-1磷酸溶液/三乙胺-乙腈两种系列作为流动相的情况下测定诺氟沙星,结果发现0.05 mol·L-1磷酸溶液/三乙胺-乙腈作为流动相时,诺氟沙星出峰时间为12.377 min,且基线不稳定;0.2%甲酸-乙腈作为流动相时,诺氟沙星出峰时间为8.533 min,
极大地节约了检测时间,且响应值较高,基线稳定,锋型更加对称尖锐,分离效果较好,信噪比更高。具体见图1、图2。此外,在试验过程中发现,荧光检测器对于甲酸与乙腈比例的变化较为敏感,梯度洗脱时引起的基线漂移较为明显,本文最终选择0.2%甲酸水溶液-乙腈(87∶13)作为流动相进行等度洗脱,洗脱时间18 min。此条件温和,不会损伤色谱柱,同时能够有效分离得到较好峰形,易控制。
2.1.2 流速优化
不同的色谱柱对同一样品有不同的保留时间,同一物质其出峰时间受流速的影响极大。一般情况下,流速由色谱柱的内径大小直接决定,内径越大流速则相对较大,样品分析时间也会相对较短,所以提高流速可大大缩短样品分析时间,但样品在色谱柱中的流速过大则会在一定程度上影响其分离效果,引起基线漂移。本试验选择1.2 mL·min-1、1.5 mL·min-1、1.8 mL·min-1 3种不同流速,观察流速对分离时间、基线和分离度的影响,结果见图3、图4和图5。
随着流速的增加,目标物出峰时间逐渐缩短,流速为1.8 mL·min-1时,出峰时间为7.285 min,峰图有稍微拖尾的现象,出峰前基线不稳现象明显;当流速为1.2 mL·min-1,出峰时间为10.643 min,目标物后含有小峰;流速为1.5 mL·min-1时出峰时间为8.530 min,峰图无拖尾现象且峰形较为尖锐对称,相对流速为1.8 mL·min-1的谱图,其基线更稳定,峰图效果好,相对流速为1.2 mL·min-1的谱图,其出峰时间有所缩短,峰图效果好,因此本试验研究最终选择流速为1.5 mL·min-1。