小麦返青期白叶性状的转录组学分析
作者: 王志龙 南文智 刘婷婷 何旭光 樊晨晨 李渊博 张雄
摘要 [目的]为解析小麦(Triticum aestivum L.)白叶突变体叶色白化机制,以白叶突变小麦体和正常小麦为试验材料,从转录水平方向展开研究。[方法]以正常小麦和温敏白叶突变体小麦为材料,进行叶色变化过程的比较转录组学分析,挖掘调控小麦光合色素形成的调控机制。[结果]参与正常小麦叶片发育过程的DGEs数目是白叶小麦叶色由白转绿过程的2.57倍,且正常叶片发育过程的DGEs主要参与脂类、氨基酸、蛋白质、嘌呤、肌醇等生物大分子的代谢,而叶色由白转绿过程的DGEs主要参与光合作用、呼吸作用和糖代谢等过程;同时发现,小麦拥有已知植物光合色素代谢途径中的所有基因,其中的75个基因为DGEs,这些DGEs包含了所有叶绿素合成途径的基因和2个类胡萝卜素合成途径的基因,这些基因的表达模式分为7类(Ⅰ-Ⅶ),表达模式表明小麦叶绿素的生物合成受反馈调节,并且叶绿素合成受阻导致试验材料形成白叶,TaCAO可能是叶绿素合成受阻的主要影响因子。[结论]该研究为进一步研究小麦叶绿素形成机制奠定了基础。
关键词 小麦;白叶性状;比较转录组学
中图分类号 Q 75 文献标识码 A 文章编号 0517-6611(2024)23-0085-09
doi:10.3969/j.issn.0517-6611.2024.23.019
Transcriptomic Analysis of White Leaf Traits in Triticum aestivum L. at Regreening Stage
WANG Zhi-long,NAN Wen-zhi,LIU Ting-ting et al
(Life Science College of Yulin University,Yulin,Shaanxi 719000)
Abstract [Objective]To decipher the mechanism of leaf color albinism in Triticum aestivum L.white-leaf mutants.[Method]This study conducted a comparative transcriptomic analysis of the leaf color change process using normal Triticum aestivumL.and temperature-sensitive white-leaf mutants.The aim was to uncover the regulatory mechanisms governing the formation of photosynthetic pigments in Triticum aestivum L..[Result]The results showed that the number of Differentially Expressed Genes (DGEs) involved in the development of normal Triticum aestivum L.leaves was 2.57 times that in the process of white leaf color turning green.DGEs in the normal leaf development process were mainly involved in the metabolism of lipids,amino acids,proteins,purines,and inositol,among other biomacromolecules.In contrast,DGEs in the process of leaf color turning from white to green were primarily involved in photosynthesis,respiration,and carbohydrate metabolism.Furthermore,it was found that Triticum aestivum L.possesses all the genes in the known plant photosynthetic pigment metabolic pathways,75 of which were DGEs.These DGEs included all genes of the chlorophyll synthesis pathway and two carotenoid synthesis pathway genes.Their expression patterns can be classified into seven categories (I to VII).These patterns suggest that the biosynthesis of Triticum aestivum L.chlorophyll is subject to feedback regulation,and that impaired chlorophyll synthesis leads to the formation of white leaves,with TaCAO likely being the main factor affecting chlorophyll synthesis.[Conclusion]These findings provide a theoretical basis for further research into the chlorophyll formation mechanism in Triticum aestivum L..
Key words Triticum aestivum L.;White-leaf trait;Comparative transcriptomics
基金项目 国家自然科学基金项目(32160462);榆林学院研究生创新基金项目(2022YLYCX08)。
作者简介 王志龙(1998—),男,陕西榆林人,硕士研究生,研究方向:作物遗传育种。*通信作者,教授,博士,硕士生导师,从事旱区农作物节水技术研究。
收稿日期 2024-01-11;修回日期 2024-02-26
叶片是植物进行光合作用的主要器官,在叶片中叶绿素是最重要的光合色素之一,类胡萝卜素是辅助光合色素,叶绿素和类胡萝卜素在植物的生物过程中有着重要的作用[1-2]。调控叶绿素和类胡萝卜素的基因发生变化往往会导致植物叶色的异常[3-4]。目前,植物合成叶绿素和类胡萝卜素的一般通路已较为明晰[5],在拟南芥中,已发现有15种酶和27个基因参与叶绿素的生物合成调控途径[6]。从细菌、真菌、藻类和植物等生物中分离出150余个调控类胡萝卜素形成的基因,它们编码类胡萝卜素合成途径中的20余种酶[7]。这些基因变异均都可能导致叶绿素和叶黄素含量发生变化[8-9]。叶色突变材料是研究光合色素生物合成、叶绿体发育以及开发基因遗传转化标记性状的理想材料[10]。
高通量测序具有测序成本低、时间短的特点[11]。这种技术可同时对数百万条序列进行测序,不仅丰富物种的遗传数据库,在功能基因的筛选和鉴定中也可以发挥作用。该技术促进了植物叶色研究领域发展,通过de novo转录组测序产生物种的遗传信息和预测可能的非编码 RNA。目前,多种叶色变异植物已被测序分析,如已经发现了许多由CHI生物合成途径突变而引起叶色变化的高等植物,如病毒诱导的CHLI(镁螯合酶I亚基)基因沉默降低了叶绿素含量并改变了叶绿体功能,导致豌豆中的叶绿体结构异常[12],在银杏中下调的HEMA会导致叶绿素积累减少[13]。然而,在小麦叶色突变体中仅报道了少数编码Mg-螯合酶D、H和I亚基的CHRD、CHLH和CHLI基因[14]。小麦中叶绿素生物合成的调控机制还有待继续深入研究。笔者选用白叶突变小麦叶色变化前后的叶片,以及同一时期叶色正常的小麦叶片,构建4个RNA文库,进行转录组测序,分析叶色变化过程中叶片基因表达谱,挖掘调控小麦叶色变白的分子机制,以期为进一步研究小麦叶色变异的分子调控机制提供参考依据。
1 材料与方法
1.1 植物材料和生长条件
试验材料于2020年10月4日种植于西北农林科技大学北校区杨凌试验站(34.16°N,108.05°E)。白叶小麦在正常温度时叶色为绿色,越冬时,低温诱导叶色变白,返青期叶色逐渐变绿。正常叶色小麦和白叶小麦分2组种植,每组3次重复,每次重复取10片叶子作为转录组混池。第1天,分别取白叶小麦的白色叶片及正常小麦的绿色叶片构建6个转录组混池;第5天,分别取白叶小麦变绿后的叶片,以及正常小麦的叶片构建6个转录组混池;共12个转录组文库。第1次取样,白叶小麦的白色叶片转录组混池记为WL,正常叶色小麦叶片的转录组混池记为GL。5 d后取样,白叶小麦转绿叶片的转录组混池记为HWL,正常叶色小麦叶片的转录组混池记为RGL。
1.2 RNA提取和Rna-Seq数据处理
利用Trizol试剂盒提取 RNA,Nanodrop 2000 对 RNA 的浓度和纯度进行检测,琼脂糖凝胶电泳检测 RNA 完整性,IN 值检测方法为 Agilent 2100 Nano,检测合格后进行文库构建和 Illumina 转录组测序,测序由百迈克生物科技有限公司完成。测序获得的原始序列 (Raw reads)去除接头(Adaptor)、含有 N(N 表示无法确定碱基信息)的比例大于 5%和低质量(质量值Q≤10 的碱基数占整个 read 的 20%以上)的数据后,得到净序列数(Clean reads)。利用Clean reads进行有参转录组分析,使用 HISAT2v2.0.5 将配对末端净序列数与小麦的参照基因组进行比对[15]。参考基因组为Triticum_aestivum.TGACv1.genome.fa。
1.3 差异表达基因、GO富集和通路分析
基因表达水平和差异基因富集分析通过featureCounts[16]计算匹配到每个基因的数据,并结合该基因的长度计算每个基因的 FPKM (reads per kilobases per million reads)值。利用 DESeq2 软件[17]对白叶小麦和正常小麦的基因进行差异表达分析,获得差异表达基因(differentially expressed genes,DEGs)。使用clusterProfiler[18]软件进行差异表达基因的 GO 和 KEGG 富集分析。
1.4 小麦光合色素相关基因的分离
利用BLAST 2.2.28+ ( ftp://ftp.ncbi.nih.gov/blast/executables/LATEST/)软件,通过其他植物中已知的调控光合色素合成的基因检索小麦基因组和转录组中的小麦同源基因。将检索得到的50分以上且匹配序列长度大于120 bp的序列保留备用,进一步利用CDD ( https:/ / www.ncbi.nlm.nih.gov / cdd / ) 数据库分析它们的保守结构域。利用 MEGA 11 软件进行小麦中叶绿素调控基因的多序列比对和进化分析,采用邻近法构建系统进化树, bootstrap为1 000。
2 结果与分析
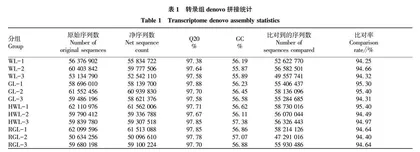
2.1 正常叶片和白叶突变体叶片发育的Rna-Seq分析
利用正常叶色小麦和白叶小麦的叶片构建了12个mRNA文库并测序,获得了50 634 256~ 62 110 976条原始序列(表1)。去除接头和低质量的序列后得到50 096 610~61 562 006条高质量序列,并且94.25%~95.40%的序列可以比对到基因组,表明测序质量和参考基因组满足高质量转录组的分析要求。