马氏珠母贝 (Pinctada fucata) 血细胞全长转录组测序与分析
作者: 王菁 李桂英 陈志炜 郭焕娣 龚晓晴 王忠良
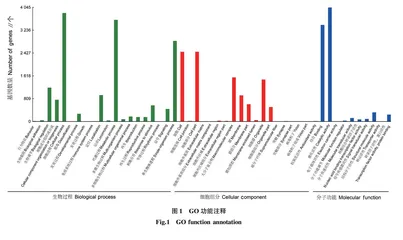
摘要 [目的]开发马氏珠母贝 (Pinctada fucata) 基因组资源,挖掘功能基因。[方法]以马氏珠母贝血细胞为试材,利用单分子实时技术(single-molecule real time,SMRT)进行全长转录组测序,并对所获得unigenes进行功能注释和基因结构分析。[结果]共获得277 064条全长非嵌合序列和82 381个基因。经过比对nr、SwissProt、KEGG 和 KOG数据库进行注释和功能分类后,共得到59 621个注释基因。同时,在8 493条基因中共发现11 219个SSR位点,其中以二核苷酸重复基元类型最高(50.9%)。生物信息学分析鉴定得到20 013个lncRNA、2 004个转录因子、10 522个可变剪切分析位点。[结论]使用SMRT技术能够深入挖掘马氏珠母贝全长转录组数据,为进一步探讨马氏珠母贝功能基因的挖掘、免疫响应及遗传机制的研究提供可靠的基因组资源。
关键词 马氏珠母贝;全长转录组;SSR标记;可变剪切
中图分类号 S 917.4 文献标识码 A
文章编号 0517-6611(2022)06-0086-05
doi:10.3969/j.issn.0517-6611.2022.06.019
开放科学(资源服务)标识码(OSID):
Full-length Transcriptome Sequencing from Hemocytes of Pinctada fucata
WANG Jing,LI Gui-ying,CHEN Zhi-wei et al (Fisheries College,Guangdong Ocean University,Zhanjiang,Guangdong 524088)
Abstract [Objective]To deep analysis of the genomic resources of the pearl oyster ( Pinctada fucata). [Method]The single-molecule real time technology (SMRT) was performed to sequence the full-length transcriptome from hemocytes,and functional annotation and the structure of the unigenes were analyzed.[Result]The results showed that a total of 277 064 full-length non-chimeric sequences were generated,and 82 381 unigenes were finally obtained.By comparing the nr,SwissProt,KEGG and KOG databases for annotation and functional classification,a total of 59 621 unigenes were annotated.GO function annotation showed that most genes were mainly enriched in cellular process,metabolic process and single biological process of biological process.Annotated genes in cell component were mainly enriched in cell and cell part,while in molecular function,they were mainly concentrated in binding and catalytic activity.Meanwhile,11 219 SSR sites were identified in 8 493 unigenes,among which the proportion of dinucleotide repeat motif was the highest (50.9%).Additionally,the results also identified 20 013 lncRNAs,2 004 transcription factors and 10 522 alternative splicing sites.[Conclusion]These results may improve knowledges on transcriptome data of P.fucata using SMRT,and provide reliable genomic resources for further exploring the development of functional genes,growth traits related gene markers,immune response and genetic mechanisms.
Key words Pinctada fucata; Full-length transcriptome;SSR;Alternative splicing
基金项目 广东省自然科学基金面上项目(2019A1515011875号); 2021年度广东省普通高校特色创新类项目(2021KTSCX044); 广东省省级科技计划项目(国际科技合作领域)(2019A050510044号);广东海洋大学2019年“冲一流”省财政专项资金建设项目;广东海洋大学2021年度本科生创新团队项目(CXTD2021001号);国家级大学生创新创业训练计划项目(CXXL2020002号); 广东海洋大学“南海学者计划”(2017年度)。
作者简介 王菁(1997—),女,蒙古族,河南南阳人,硕士研究生,研究方向:水产经济动物生物学。*通信作者,副教授,从事水产经济动物功能基因与基因组学研究。
收稿日期 2021-10-29
马氏珠母贝 (Pinctada fucata) 是我国海水珍珠养殖的主要母贝,主要分布于我国南海沿岸[1]。目前,关于马氏珠母贝的研究普遍集中于生长性状[2-3]、矿化功能[4]、免疫响应[5]等方面。分析不同性状形成的机理、性状相关基因分子标记的开发以及遗传机制的研究,都需要借助于马氏珠母贝的基因组资源[6]。随着基因组和转录组数据资源的不断开发,全长转录组测序技术能够提高转录组注释的准确性,提供一种更加全面的功能基因注释方法[7]。转录组测序不仅可以反映细胞内基因的类型和数量,还可以在分子水平上揭示细胞的生理生化过程[8],具有识别参与机体生长、发育和免疫调节等生物过程的相关基因以及分析基因表达调控的重要功能[9]。第三代测序技术PacBio利用单分子实时测序技术(single-molecule real time,SMRT)直接测序,无需经过组装过程即可直接生成包含5’-UTR、3’-UTR和poly-A尾巴的完整转录本[10]。相较于第二代测序技术,PacBio平台所得到的转录本更长,转录本注释率也得到提高[11]。使用SMRT技术对花鲈 (Lateolabrax maculatus) 进行测序分析增加了对硬骨鱼类盐度适应机制的理解[12]。Katneni等[7]对印度对虾 (Penaeus indicus) 全长转录组数据进行分析,为进一步研究相关经济性状的功能基因提供宝贵的基因组资源。使用SMRT技术对金乌贼 (Sepia esculenta) 进行测序并获得高质量的数据库,为探索生长发育、组织分化等相关性状提供了基础资源[13]。Chen等[9]对达氏鲟 (Acipenser dabryanus) 进行全长转录组测序,鉴定得到3个 TRIM 基因,并证明其在抗细菌感染的免疫反应中起着关键作用。Liu等[14]对平胸龟 (Platysternon megacephalum) 肝脏进行单分子实时测序,系统分析线粒体DNA转录组的基因重排和控制区重复线性现象,加深了对线粒体基因组转录调控机制的理解。此外,全长转录组数据可用于分析选择性剪接事件(alternative splicing,AS),有助于进一步理解RNA的加工过程[6,15],而综合分析转录组基因的剪接异构体,有助于完善现有基因模型的功能注释[12]。
该研究使用SMRT技术对马氏珠母贝血细胞进行全长转录组测序、功能基因挖掘、可变剪切分析,旨在提高现有马氏珠母贝基因组资源功能注释的准确性及多样性,对于进一步开展马氏珠母贝功能基因的挖掘及其免疫机制的研究提供参考。
1 材料与方法
1.1 马氏珠母贝的暂养及总RNA提取
马氏珠母贝(壳长约65 mm)购自广东省湛江市雷州养殖场,暂养于室内海水桶,1 d换水1次;水温25 ℃,暂养期间保持充气,定时投喂螺旋藻粉。室内暂养7 d后随机选择3个马氏珠母贝于闭壳肌处采集血淋巴,混合为1个样品,4 ℃、1 200 r/min条件下离心10 min收集血细胞,进一步提取总RNA及进行全长转录组测序。
1.2 全长转录组文库构建及测序
按照制造商说明书进行SMRTbell文库构建,采用MagBead Loading上机测序获得原始数据,使用SMRT Link v6.0对原始数据进行分析提取CCS(Circular Consensus Sequence)序列,用ICE (Interactive Clustering and Error Correction)算法和Quiver算法对FLNC(full-length non-chimeric)进行聚类和校正,得到的全长转录组数据已提交至NCBI数据库(SRR9644402)。
1.3 基因比对及功能注释
Unigene在Nt、Nr、Swiss-Prot、KEGG、COG 及Pfam 数据库中比对并获得基因注释信息( E 值<1×10-5)。使用Blast2GO软件得到序列的 GO 注释信息,根据 KEGG 注释信息进一步得到通路的注释信息,利用iTAK软件预测转录因子(TF)。
1.4 SSR、lncRNA及可变剪切分析
使用MISA(http:∥pgrc.ipk-gatersleben.de/misa/)软件对转录组数据中所有unigenes进行检索并鉴定unigenes中的串联重复单元(Simple sequence repeat,SSR)。对未注释到数据库的全长序列进行lncRNA分析,使用cnci软件[16]和CPC软件[17]进行编码能力预测得到lncRNA。使用Cogent[18]软件组装出编码序列,然后以组装好的序列作为参考,使用SUPPA[19]软件进行可变剪切分析。
1.5 RT-PCR 验证 为了验证通过SMRT技术获得的转录本准确性,提取马氏珠母贝血细胞总RNA,反转录为cDNA第一链,基于SMRT获得的unigenes中随机选择5个基因进行特异性引物设计(表1),使用RT-PCR及产物回收送至上海生工生物技术公司进行测序。
2 结果与分析
2.1 SMRT测序数据统计
利用SMRT测序技术,共获得7 640 496个subreads,平均长度为1 838 bp,N50为2 563 bp(表1)。使用Permissive CCS2软件进行序列提取和质量过滤后,共得到346 737条循环一致序列(CCS)。其中277 064条为全长非嵌合序列(FLNC),平均长度为2 296 bp。对上述FLNC进行聚类和校正后,共获得169 961条一致性序列(ICE consensus),平均长度为2 384 bp。序列去冗余后,最终获得82 381条校正序列(unigenes),平均长度为2 512 bp,N50为2 861 bp(表1)。