反相高效液相色谱法测定牛初乳保健食品中免疫球蛋白G的含量
作者: 罗娇依 刘彤彤 曹进 赵溪 孙姗姗 张旭光
摘 要:目的:建立一种测定牛初乳保健食品中免疫球蛋白G的反相高效液相色谱法。方法:样品采用磷酸盐缓冲液涡旋振荡提取,经Protein G固相免疫亲和小柱净化后,使用高效液相色谱仪(搭载紫外检测器)检测,紫外波长为280 nm,采用C4反相色谱柱(4.6×250 mm,5 μm)分析。流动相为0.1%三氟乙酸-水溶液和乙腈,梯度洗脱分离,外标法定量。结果:色谱分离过程可在10 min内完成,免疫球蛋白G在10~100 μg/mL范围内线性良好,相关系数R=0.999 6。检出限为0.5 g/kg,定量限为1.7 g/kg。3个浓度水平的方法加标回收率为92.2~97.5%,方法精密度为0.20~1.18%。样品经前处理后4 ℃冰箱内保存,在24 h内稳定性良好。结论:该方法特异性强,灵敏度、精确度高,符合方法学验证要求,可快速、准确地对保健食品中免疫球蛋白G进行分析检测。
关键词:免疫球蛋白G;保健食品;高效液相色谱法;Protein G免疫亲和柱
免疫球蛋白(Ig)按其抗原性不同可分为5类:IgG、IgA、IgE、IgM和IgD[1]。其中,免疫球蛋白G(IgG)是最主要的免疫因子,并且在人体内占总血清免疫球蛋白含量的70%~80%左右[2]。IgG是一种母体给幼崽传递特异性免疫力的主要载体,其具有抗原结合和凝集特性,具有中病原微生物(如病毒)和毒素的作用[3]。其他研究表明,IgG对人体和犊牛的免疫功能有有益影响,如防止腹泻、促进非特异性免疫、增强细胞免疫功能、体液免疫功能等[4-6]。因IgG特有的增强免疫功能,近些年,在普通食品的研发上也受到了关注,如富含免疫球蛋白的冰淇淋、臻糕、发酵乳等[7-9]。而与常乳相比较,牛初乳中IgG等涉及物质转运和免疫活性等功能的蛋白质表达量更高[10-11],且IgG含量高低也作为牛初乳产品生产工艺的优劣评判标杆[12]。所以,在保健食品中,以IgG为主要营养成分的产品多以牛初乳为原料。
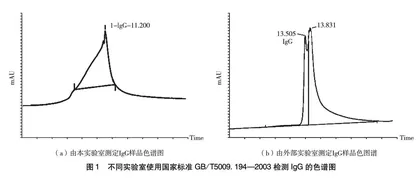
目前,现有国家标准测定IgG的方法有酶联免疫吸附法、琼脂单向免疫扩散法、分光光度法、高效亲和色谱法(HPAC)等[13-16]。牛初乳保健食品检测IgG最常见的方法为国家标准GB/T 5009.194—2003,原理基于HPAC,其将IgG亲和柱(Pharmacia HI-Trap Protein G柱,1 mL)适配在高效液相色谱上,利用IgG的特异吸附性作用对样品进行在线检测[17]。IgG存在多种亚型,如IgG1、IgG2、IgG3、IgG4,在牛初乳中富含IgG1和IgG2[18-19]。因此,在使用GB/T5009.194—2003检测时,亲和吸附后洗脱解吸附时会造成IgG混合峰型分叉,较大峰宽,前延、拖尾明显,且无法与其他杂质峰分开(图1),造成积分困难,导致无法准确定量。因此,为了提高结果精确度,现开发一种对保健食品中IgG含量测定的高效液相色谱法,以确保牛初乳原料的质量控制以及牛初乳保健食品中功效成分含量的精准测定,为企业提供检测依据,为保健食品品质监管提供有利技术手段。
1 材料与方法
1.1 材料与仪器
牛血清IgG标准品(纯度≥95%),由Sigma提供(货号:I5506);牛免疫球蛋白G亲和柱3 mL,由美正生物提供(货号:QC0137);三氟乙酸(色谱纯),Alfa Aesar提供;乙腈(质谱纯),Fishers提供;磷酸盐缓冲液片剂(PBS)pH=7.2~7.4,Salarbio提供;GHP滤膜(0.22 μm),Waters公司提供;实验室用水为Milli-Q超纯水;含IgG的保健食品样品为市场购买和汤臣倍健股份有限公司提供。
U3000高效液相色谱仪(配有紫外检测器及Chromeleon数据处理系统),美国赛默飞公司;AL204和XP205电子分析天平,梅特勒-托利多公司;CR22GIII高速离心机,日本日立公司。
1.2 方法
1.2.1 IgG提取工艺 相较于国标法中直接采用流动相稀释样品后分析,本方法认为需考察样品前处理的提取方法,最大化的回收待测成分以满足检测方法学的要求。但牛乳类基质中都会含有IgG本底值,无法获得与牛乳相似的混合性蛋白溶液作为阴性基质提取效率的考察对象,故只能选择使用同一样品制得的平行样品,分别比较直接稀释溶解法(记为0 min)与涡旋振荡法、超声法的提取效率。提取溶液选择pH=7.2~7.4的0.1 mol/L PBS溶液,可保证免疫亲合柱抗原、抗体之间较强的作用力和较好的净化效果[20]。其中振荡法考察了1、3、5 min的时间节点(n=3),而超声法选择的时间节点为5、10、20 min(n=3)。
1.2.2 标准溶液的配制 标准储备液的制备:精密称取牛血清免疫球蛋白G标准品适量(精确至0.01 mg),置棕色容量瓶中,用0.1 mol/L PBS溶解并制成每1 mg /mL的IgG储备溶液,摇匀,即得。标准系列工作液的制备:分别准确吸取不同体积的标准储备液,加0.1 mol/L PBS将其稀释成IgG含量分别为10、20、40、60、80、100 μg/mL的标准系列工作液。临用时配制,冷藏。
1.2.3 样品的前处理 将样品混合均匀后,精密称取混匀试样约0.100 0 g,置50 mL具塞离心管中,用0.1 mol/L PBS定容至刻度,充分振荡混匀1 min后,8 000 r/min离心5 min,取上清液5 mL过牛免疫球蛋白G亲和柱,最终使用洗脱液定容至5 mL,取部分洗脱液过0.22 μm滤膜,得滤液,即可上机。
1.2.4 色谱条件 本方法采用的反相高效液相色谱法检测。色谱柱:沃特世 Symmetry300 C4色谱柱(4.6 × 250 mm,5 μm);流动相:0.1%三氟乙酸-水溶液和乙腈,流动相梯度见表1;紫外检测波长:280 nm[21];柱温30 ℃;进样体积20 μL。
1.3 方法学考察
1.3.1 标准曲线、检出限和定量限 将标准系列工作液检测的峰面积为Y轴、标准溶液浓度为X轴,进行线性回归分析,得线性方程和相关系数。在检测检出限和定量限时,经检测多个乳粉样本以及保健食品样品(复合蛋白粉)都发现含有少量的IgG本底值,无法采用不含本底的阴性基质加标的方式精准地确定本方法的检出限与定量限。因此,选取与乳粉相似的多蛋白混合豆粉作为阴性基质,采用逐步添加IgG标准溶液的方式来确定检出限和定量限。
1.3.2 方法回收率和精密度 在研究本方法回收率与精密度时采用的是同一保健品样品进行含本底值的加标实验。根据线性范围以及样品真实含量情况,制定了3个水平添加浓度,每个浓度取6份平行样(n=6),为保证高浓度加标样的测定浓度在线性范围内,将洗脱液最终定容至10 mL。
1.3.3 样品稳定性 由于IgG为生物活性物质,在分析过程中的前处理操作温度尽可能低以及减少处理时间才可最大化保持其生物结构,该结构的保持直接关系到在280 nm处的波长吸收效率。为考察样品24 h内的稳定性情况,将两种剂型样品经前处理后,使用封口膜密封并贮存于4 ℃冰箱内。在冷藏后,于0、0.5、1、2、4、8、12、24 h等时间点,对样品分别进行检测,分析稳定性。
1.4 IgG的含量计算
将标准工作液和制备后的样品,参照仪器条件检测,将样品检测结果带入标准曲线,按照式(1)计算,即得最终含量。
X=n×V×fm×1 000×1 000(1)
式(1)中,X为试样中 IgG的含量(g/100 g);n为试样中 IgG的质量浓度(μg /mL);m为试样的取样量(g);V为试样的定容体积(mL);f 为样液稀释因子;100、1 000为换算系数。
2 结果与分析
2.1 前处理方法的分析
2.1.1 提取方法和时间的优化 如图2所示,经one-way ANOVA统计分析,与直接稀释溶解相比,振荡法和超声提取法均有显著增强(P≤0.05)。但振荡法的提取效率在1 min后无显著性增强(P≥0.05),则以1 min为振荡法的最佳选择。超声法结果均无显著增高于涡旋振荡法1 min结果。则经考虑对比实验时间消耗和样品含量回收的增幅比例,在相似提取效率的结果下,涡旋振荡1 min为此方法样品前处理的优选提取条件。
2.1.2 净化方法的分析 在传统的IgG工业纯化方法中,因为需要经过化学沉淀、透析、浓缩等多个分离步骤,导致在生产过程中IgG的大量损失[22],因此本研究采用特异性吸附能力更强的免疫亲和柱法作为净化免疫球蛋白G的方法。Protein G免疫亲和柱填料作为强力吸附乳制品或者血液样品中IgG的手段已经被多次报道,并已形成商品化的预充柱[23-24]。相比Protein A填料,Protein G填料对牛源IgG结合能力更强,可作用于全部IgG亚型,并不会作用于其他免疫球蛋白[25]。国标GB/T 5009.194—2003中规定了该种填料的预充柱作为液相色谱分离过程中的分析柱使用,但是经实际样品检测过程中发现,此预充柱并非适配于绝大多数液相色谱仪型号,需增加较多串口配件;在适配过程中对于系统压力的要求较高,常出现超压漏液等现象,不易控制;分离流动相系统包含了磷酸盐以及甘氨酸盐酸盐,此类洗脱液极易造成液相系统管路内产生结晶、堵塞,不利于仪器维护。本实验将这种在线分离方式转化成为柱前分离净化的方式操作。采用含Protein G的固相萃取小柱,将IgG提取溶液预先通过亲和小柱进行吸附与解吸附的处理,最大化地去除在280 nm处与IgG共流出峰的干扰。如图3所示,a为标准品色谱图,b、c为2种剂型的牛初乳保健食品样品经前处理提取和净化后测定的IgG液相色谱图,待测峰均呈现了良好的分离情况,且峰型尖锐对称,10 min内即可获得完整的IgG色谱峰。比较图3中的总分离时间段内各色谱图可发现,经固相萃取柱净化后的不同剂型样品色谱图几乎均与标准品色谱图一致,且提高了对IgG的净化效率。可以有助于计算方法的回收效率,避免亲和柱过载时导致的在线分析干扰,优化了分离柱上的峰型,使积分准确。
2.2 方法学考察
2.2.1 标准曲线、检出限与定量限的确定 采用峰面积与分析物浓度进行线性回归计算,得到线性方程为Y=0.003 4X+0.011 4,相关系数R为0.999 6。因此,IgG液相分析法在10~100 μg/mL的浓度范围内线性呈良好关系。IgG色谱峰信噪比约等于 3 时的浓度作为检出浓度,为1.0 μg/mL;以及信噪比约于10 时的浓度作为定量浓度,为3.3 μg/mL。则此方法的方法检出限和定量限分别为0.5、1.7 g/kg。
2.2.2 回收率与精密度的验证 3个浓度样品经检测结果计算得,低浓度加标回收率为97.5%,中浓度加标回收率为92.2%,高浓度加标回收率为93.5%,精密度结果为0.20%~1.18%(表3)。回收率和精密度均符合方法学验证要求。
2.2.3 样品稳定性的评价 将经前处理后的2种剂型样品于4 ℃环境中冷藏贮存24 h,并监测24 h内各时间节点的样品IgG含量以考察稳定性。如表4所示,24 h内样品中IgG的浓度值的精密度为1.04%~1.72%,偏差较小,表明样品经前处理后,24 h内稳定。
3 市售牛初乳保健食品样品检测
共收集市售的经保健食品注册备案的牛初乳保健食品样品15批,编号为1~15号。其中8号样品为进口产品,其余均为国产产品,声称的功效成分均为“免疫球蛋白G”(其中9号样品声称功效成分为“免疫球蛋白”)。使用本方法和国标法(GB/T 5009.194—2003)同时测定这15批样品,对结果进行比较,检测结果如表5所示。根据GB 13432规定,营养成分的实际含量不应低于标示值的80%[26],1号样品2个方法的检测结果都低于标示值的80%,8号样品的本方法检测结果低于标示值的80%,其他产品2个方法的检测结果均高于标示值的80%。在检测过程中发现,方法空白在国标法中会在IgG出峰时间引入杂质峰(图4),此杂质峰相当于约5 g/100 g的IgG浓度,则在检测低IgG含量的样品检测时,如6号、8号、10号样品,无法保证检测结果的准确性。与本方法的含量结果对比,国标法检测的8号样品含量结果高于标示值,可能是杂质峰的引入导致,无法用来判断产品是否合格。