基于机器学习的蛋白质亚细胞定位预测方法
作者: 李佳楠 李卓 滕小华 高兴泉 唐友
摘要 蛋白质亚细胞定位预测有助于了解蛋白质性质和功能,理解蛋白质复杂的生理过程和调控机理,对开发新药物等方面有着很大的促进作用。随着人类在蛋白质组学方面研究的不断深入,蛋白质序列数据量呈指数式增长,单纯依靠传统的试验方法已无法满足生命科学研究的需要,于是人们将蛋白质亚细胞定位的研究方法转向了机器学习领域,研究机器学习算法变得越来越重要。从蛋白质序列特征的刻画、预测算法、算法评价3个方面阐述现阶段蛋白质亚细胞定位预测的研究进展。最后,总结蛋白质亚细胞定位预测方法方面取得的成果及需要不断完善的3个方面(特征选择、数据处理和改进算法),并提出了未来机器学习在提高预测性能方面的研究重点及重要意义。
关键词 蛋白质;亚细胞定位;机器学习;预测算法
中图分类号 TP 181 文献标识码 A
文章编号 0517-6611(2022)16-0198-07
doi:10.3969/j.issn.0517-6611.2022.16.048
开放科学(资源服务)标识码(OSID):
Prediction Method of Protein Subcellular Localization Based on Machine Learning
LI Jia-nan1,LI Zhuo2,TENG Xiao-hua2 et al
(1.School of Information and Control Engineering, Jilin Institute of Chemical Technology, Jilin, Jilin 132000; 2.Electrical and Information Engineering College, Jilin Agricultural Science and Technology University, Jilin, Jilin 132101)
Abstract Prediction of protein subcellular localization helps to understand the properties and functions of proteins, to understand the complex physiological processes and regulatory mechanisms of proteins, and has a great role in promoting the development of new drugs.With the continuous deepening of human research in proteomics, the amount of protein sequence data has increased exponentially.Relying solely on traditional experimental methods can no longer meet the needs of life science research, so people have turned to the research methods of protein subcellular localization.In the field of machine learning, research on machine learning algorithms is becoming more and more important.We described the current research progress of protein subcellular location prediction from three aspects of characterization of protein sequence features, prediction algorithmsand algorithm evaluation.Finally, we summarized the achievements of protein subcellular localization prediction methods and the three aspects that need to be continuously improved, which were feature selection, data processing and improved algorithms.The research focus and important significance of future machine learning in improving prediction performance were put forward.
Key words Protein;Subcellular localization;Machine learning;Prediction algorithm
蛋白质的亚细胞定位与其功能紧密相关,蛋白质只有处于正确的亚细胞位置才能维持细胞系统的正常运转,蛋白质亚细胞定位研究不仅能够帮助人们了解蛋白质的性质和功能和蛋白质之间的调控机制,还能为人们开发新药物提供有效的参考信息[1]。大多数蛋白质只能在细胞中的1个特定位置(如细胞核、细胞膜)发挥作用,然而一些其他的蛋白质可以在细胞中的几个位置发挥作用[2]。一个蛋白质想要正常的发挥功能,必须处于细胞中的一个或几个特定的位置上,否则该蛋白质就会失效[3]。自后基因组时代以来,产生了大量的蛋白质序列,单纯依靠传统的实验方法进行蛋白质亚细胞定位十分的耗时、耗力[4]。为了更加精准、快速解决蛋白质亚细胞定位问题,人们将研究方法从传统的实验手段逐步扩展到机器学习领域。鉴于此,笔者从蛋白质序列特征的刻画、预测算法、算法评价3个方面阐述现阶段蛋白质亚细胞定位预测的研究进展,总结蛋白质亚细胞定位预测方法方面取得的成果及需要不断完善的3个方面(特征选择、数据处理和改进算法),并提出了未来机器学习在提高预测性能方面的研究重点及重要意义。
1 蛋白质序列特征刻画
对国内外相关研究的分析显示,机器学习领域的蛋白质亚细胞定位的发展基本可以分为5个阶段:第1阶段(2006—2010年)的工作主要集中在预测单位点的蛋白质亚细胞位置,但忽略了多位点蛋白质的存在[5]。第2阶段(2011—2013年)的工作主要集中在单位点和多位点蛋白质亚细胞位置的预测[6],但是大部分为多位点蛋白质开发的技术,在尝试进行预测时却将多位点的问题转化为了单位点的问题。第3阶段(2017—2018年)使用不同的特征提取技术,例如将基因本体(GO)信息融合到通用伪氨基酸组成(PseAAC)中,为多标签蛋白质亚细胞定位开发出了许多的预测器[7]。第4阶段(2018—2020年)是在用预测器对特征提取后的特征向量进行预测之前,使用不同的数据平衡技术处理多标签蛋白质亚细胞定位中的数据不平衡问题[8]。第5阶段(2020—2021年)通过优化机器学习算法以及特征融合来提升预测的准确性,其中具有代表性的算法有深度学习和集成学习[9]。
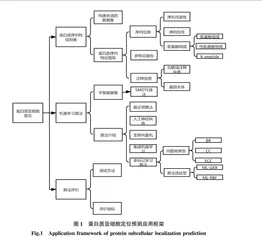
很多研究者[10-11]在阐述关于蛋白质亚细胞定位的相关研究时,都用到了Chou[12]的五步法则:①有效构建优质的基准数据集用于模型/分类器的训练与预测;②从蛋白质样本中提取可用于区分不同类别的蛋白质的相关特征;③采用或设计1个优异的分类算法,用于预测各自类别中的不同蛋白质;④选择1个合适的验证方法直观的评价分类模型的有效性;⑤构建1个可公开访问的用户友好型的网络服务器。具体机器学习方法在蛋白质亚细胞定位预测中的应用框架如图1所示。
在进行蛋白质序列特征刻画之前需要构建一个合适的数据集,数据集是算法模型训练和测试的数据基础,它决定了模型训练和测试的效果,因此构建一个合适的数据集十分重要。在构建数据集时应考虑到以下5个因素:①蛋白质序列条数;②需要预测的位点的个数;③是否需要研究多位点定位问题;④特定物种数据集以及基因组数据集的差异;⑤序列同源性大小控制。
目前使用的数据集基本来源于Swiss-Prot[13]数据库和其他的一些关于物种和位置的专门的数据库,如PPDB[14](plant proteomics database)和NPD[15](nuclear protein data base)等。
蛋白质是由氨基酸组成的,蛋白质组成形式可由如下公式表示:
P=R 1R 2…R n(1)
式中,P代表蛋白质序列,R i(i=1,2,…,n)代表蛋白质序列下的每一个氨基酸。
蛋白质序列原始字母式数据无法直接经过机器学习的方法进行分类和处理,因此需要先将蛋白质的数据转换为一种能够准确地刻画出序列模式信息的离散性数据,再通过机器学习的算法对其进行接下来的分类和处理操作[16]。20种不同的氨基酸残基按照不同的排列组合形成了蛋白质序列,序列中包含了进化特征、序列特征、理化特征等,这些特征对算法的设计和预测结果都会产生影响。提取的特征过少会导致提取后的数据缺失一些重要信息,影响最终预测的结果;提取的特征过多则会导致维数灾难[17],严重影响算法的效率。因此,如何提取有效的特征并进行融合来提升算法预测的结果仍然是现阶段的核心问题。该研究从序列信息、注释信息和多特征融合3个方面来介绍目前主要使用的特征提取方法:
1.1 序列信息 基于序列信息进行蛋白质序列特征提取的表示方法又可细分为以下3种方法:序列同源性、序列信号、氨基酸组成[18]。
(1)序列同源性。基于序列同源性方法[19]主要通过一些相似性比对工具进行序列间的相似性检验:BLAST(Basic local alignment search tool)、PSI-BLAST是2个很常用的相似性比对搜索工具,PSI-BLAST在BLAST的基础上做了一定的改进,改良过后的PSI-BLAST可对同源性较低的序列之间进行相似性度量。2005年Xie等[20]、2006年Guo等[21]将蛋白质序列同源性信息用于蛋白质亚细胞定位,该方法的缺点为对于一些待测的蛋白质,并不能找到同源性较高的蛋白质序列与之匹配,那么该方法将不再有效。
(2)序列信号。蛋白质的序列上拥有着一部分特殊的子序列,同样特殊的子序列位于蛋白质的N端,而此类子序列被称为分选信号。分选信号的存在会使的蛋白质在功能开展及分选过程当中,转移到特定的亚细胞的位置。目前,已知的分选信号有信号肽、叶绿体运输肽、线粒体转移肽等。序列信号的研究工作一直持续进行,并取得了一定的研究成果,如2000年Emanuelsson等[22]利用N端分选信息预测叶绿体运输肽;2007年Emanuelsson 等[23]开发了基于N端分选信号的蛋白质亚细胞定位方法;2012年Tardif等[24]基于N端分选信号开发了可进行绿藻亚细胞定位预测的工具:PredAlgo。
(3)氨基酸组成。氨基酸是蛋白质序列当中简单直接的特征。ACC的向量表示形式为:
X=[x 1,x 2,x 3,…,x n](2)
式中,x i(i=1,2,3,…,n)表示蛋白质在X中的20中原生氨基酸出现的频率。1994年,Nakashima等[25]最早利用组成蛋白质氨基酸含量的百分率来区分细胞内和细胞外的蛋白质;1995年Chou[26]对ACC的表现形式由原本的20维简化至19维,发现两者是等价的;1998年Reinhardt等[27]在Nakashima和Nishikawa的基础上提出了用氨基酸对进行蛋白质亚细胞定位,构造了蛋白质亚细胞定位第1个人工神经网络。在接下来的几年里,ACC在蛋白质亚细胞定位领域得到了广泛的使用[28]。该方法的缺点为氨基酸组分无法反应序列的局部信息,只能反应序列的整体信息,且氨基酸组分的方法未能考虑到氨基酸的物理化学性质,因此氨基酸组分具有局限性。2000年Chou[29]将序列的顺序因素加入氨基酸组成中进行蛋白质亚细胞定位,发现该方法能有效地提升最终预测结果。